Składniki aktywne: Erytropoetyna (epoetyna alfa)
EPREX 2000 j.m./ml, 4000 j.m./ml, 10 000 j.m./ml i 40 000 j.m./ml ROZTWÓR DO WSTRZYKIWAŃ W AMPUŁKO-STRZYKAWKACH
Dlaczego stosuje się Eprex? Po co to jest?
Co to jest EPREX i do czego służy
EPREX zawiera substancję czynną epoetynę alfa, białko, które pobudza szpik kostny do wytwarzania większej liczby czerwonych krwinek, komórek przenoszących hemoglobinę (substancję zdolną do przenoszenia tlenu).Epoetyna alfa jest kopią ludzkiej erytropoetyny i działa w ten sposób.
- EPREX stosuje się w leczeniu objawowej niedokrwistości spowodowanej niewydolnością nerek.
- u dzieci poddawanych hemodializie
- u dorosłych poddawanych hemodializie i dializie otrzewnowej
- u osób dorosłych z ciężką niedokrwistością niepoddawanych jeszcze dializie.
Jeśli u pacjenta występuje niewydolność nerek, a nerki nie wytwarzają wystarczającej ilości erytropoetyny (niezbędnej do wytwarzania czerwonych krwinek), we krwi może być niewiele czerwonych krwinek. EPREX jest przepisywany w celu stymulowania szpiku kostnego do wytwarzania większej liczby czerwonych krwinek.
- EPREX jest stosowany w leczeniu anemii, która może wystąpić podczas chemioterapii guzów litych, chłoniaka złośliwego lub szpiczaka mnogiego (raka szpiku kostnego), jeśli lekarz uzna, że może być konieczna transfuzja krwi.EPREX może zmniejszyć potrzebę transfuzji.
- EPREX stosuje się w leczeniu pacjentów z umiarkowaną niedokrwistością, którzy są kandydatami do odkładania krwi w oczekiwaniu na operację, aby można ją było przetoczyć w trakcie lub po operacji.Ponieważ EPREX stymuluje wytwarzanie czerwonych krwinek, możliwe jest pobranie krwi od tych osób większa ilość krwi.
- Preparat EPREX stosuje się w leczeniu dorosłych pacjentów z umiarkowaną niedokrwistością poddawanych poważnym operacjom ortopedycznym (np. alloplastyka stawu biodrowego lub kolanowego) w celu zmniejszenia potrzeby transfuzji krwi.
Przeciwwskazania Kiedy nie należy stosować leku Eprex
Nie stosować EPREX
- Jeśli u pacjenta stwierdzono uczulenie (nadwrażliwość) na epoetynę alfa lub którykolwiek z pozostałych składników leku EPREX (wymienionych w punkcie Zawartość opakowania i inne informacje);
- Jeśli po uprzednim leczeniu jakąkolwiek substancją stymulującą wytwarzanie krwinek czerwonych (w tym EPREX) zdiagnozowano „czystą aplazję czerwonokrwinkową” (szpik kostny nie może wytworzyć wystarczającej ilości czerwonych krwinek). Patrz punkt „Możliwe działania niepożądane”.
- Jeśli masz problemy z niekontrolowanym nadciśnieniem tętniczym
- Stymulowanie produkcji czerwonych krwinek (aby można było pobrać więcej krwi), jeśli pacjent nie może otrzymać transfuzji własnej krwi w trakcie lub po zabiegu.
- Jeśli masz przejść planową dużą operację ortopedyczną (np. endoprotezoplastykę stawu biodrowego lub kolanowego) oraz:
- ma ciężką chorobę serca
- masz poważne problemy z żyłami i tętnicami
- niedawno miał zawał serca lub udar mózgu
- nie może przyjmować leków rozrzedzających krew
EPREX może nie być odpowiedni dla Ciebie.Prosimy omówić to z lekarzem. Podczas stosowania leku EPREX niektóre osoby mogą potrzebować leków zmniejszających ryzyko powstawania zakrzepów krwi.Jeśli pacjent nie może przyjmować leków przeciwzakrzepowych, nie powinien przyjmować leku EPREX.
Środki ostrożności dotyczące stosowania Informacje ważne przed przyjęciem leku Eprex
Zachowaj szczególną ostrożność z EPREX
EPREX i inne środki stymulujące czerwone krwinki mogą zwiększać ryzyko powstania zakrzepów krwi u wszystkich pacjentów. Ryzyko to może być większe, jeśli u pacjenta występują inne czynniki ryzyka powstania zakrzepów krwi (na przykład, jeśli miałeś zakrzepy krwi w przeszłości lub masz nadwagę, masz cukrzycę, chorobę serca lub jesteś unieruchomiony przez długi czas z powodu operacja lub choroba). Powiedz swojemu lekarzowi o którejkolwiek z tych rzeczy. Lekarz pomoże Ci zdecydować, czy EPREX jest dla Ciebie odpowiedni.
Ważne jest, aby porozmawiać z lekarzem, jeśli którakolwiek z poniższych sytuacji dotyczy Ciebie.
Nadal można stosować EPREX, ale należy to najpierw omówić z lekarzem.
- Jeśli wiesz, że cierpisz lub cierpiałeś na:
- Wysokie ciśnienie krwi;
- Napady lub napady
- Choroba wątroby;
- niedokrwistość z innych przyczyn;
- Porfiria (rzadka choroba krwi).
- Jeśli masz raka, powinieneś mieć świadomość, że substancje stymulujące produkcję czerwonych krwinek (takie jak EPREX) mogą działać jako czynnik wzrostu i teoretycznie wpływać na progresję nowotworu. Transfuzja krwi może być preferowana w zależności od indywidualnego stanu. Porozmawiaj o tym z lekarzem.
Zwróć szczególną uwagę na substancje stymulujące produkcję czerwonych krwinek:
EPREX należy do grupy substancji stymulujących produkcję czerwonych krwinek, takich jak ludzkie czynniki erytropoetyczne. Lekarz zawsze dopilnuje, aby zapisać dokładną nazwę produktu, którego używa. W przypadku podania w trakcie leczenia substancji należącej do tej samej grupy, ale innej niż EPREX, przed zastosowaniem należy skontaktować się z lekarzem lub farmaceutą.
Interakcje Jakie leki lub pokarmy mogą zmienić działanie leku Eprex
EPREX zwykle nie koliduje z innymi lekami, ale zawsze należy poinformować lekarza, jeśli pacjent przyjmuje lub ostatnio przyjmował jakiekolwiek inne leki, w tym te, które nie wymagają recepty.
Jeśli pacjent przyjmuje lek o nazwie cyklosporyna (stosowany na przykład po przeszczepieniu nerki), lekarz może zlecić badania krwi w celu sprawdzenia stężenia cyklosporyny podczas leczenia lekiem EPREX.
Suplementy żelaza i inne czynniki zapobiegające anemii mogą zwiększyć skuteczność leku EPREX.Twój lekarz zdecyduje, czy należy je przyjmować.
W przypadku hospitalizacji lub badania lekarskiego prosimy o poinformowanie, że jest Pani leczona preparatem EPREX. Może to wpłynąć na inne zabiegi lub wyniki testów.
Ostrzeżenia Ważne jest, aby wiedzieć, że:
Ciąża i karmienie piersią
Ważne jest, aby poinformować lekarza, jeśli którykolwiek z poniższych stanów dotyczy Ciebie. Nadal możesz stosować EPREX, ale najpierw porozmawiaj o tym z lekarzem.
- Jeśli jesteś w ciąży lub myślisz, że możesz być.
- Jeśli karmisz piersią.
Dawka, sposób i czas podawania Jak stosować Eprex: dawkowanie
Ten lek należy zawsze stosować zgodnie z zaleceniami lekarza. Skontaktuj się z lekarzem, jeśli nie masz pewności.
Na podstawie badań krwi lekarz ustalił, że konieczne jest stosowanie leku EPREX.
EPREX można podawać we wstrzyknięciu:
- Do żyły lub rurki do żyły (dożylnie)
- Pod skórą (podskórnie)
Lekarz zadecyduje o sposobie podawania leku EPREX. Zastrzyki są zwykle podawane przez lekarza, pielęgniarkę lub pracownika służby zdrowia; niektóre osoby mogą nauczyć się samodzielnie podawać lek podskórnie: patrz Instrukcja samodzielnego wstrzykiwania leku EPREX.
- Eprex nie powinien być stosowany:
- po upływie daty ważności wskazanej na etykiecie lub opakowaniu zewnętrznym
- jeśli wiesz lub myślisz, że lek mógł zostać przypadkowo zamrożony lub tak
- i była awaria lodówki
Dawka leku EPREX jest ustalana na podstawie masy ciała w kilogramach i zostanie dobrana przez lekarza w zależności od przyczyny niedokrwistości.
Lekarz będzie regularnie kontrolował ciśnienie krwi podczas leczenia lekiem EPREX.
Pacjenci z niewydolnością nerek
- Twoja wartość hemoglobiny będzie utrzymywana między 10 a 12 g / dl, ponieważ wyższe poziomy hemoglobiny mogą zwiększać ryzyko zakrzepicy i śmierci.
- Zazwyczaj stosowana dawka początkowa leku EPREX dla dorosłych lub dzieci to 50 jednostek międzynarodowych (j.m.) na kilogram (/kg) masy ciała, podawana 3 razy w tygodniu.
- U pacjentów poddawanych dializie otrzewnowej podawanie można wykonywać dwa razy w tygodniu.
- EPREX podaje się dożylnie (żyła lub rurka do żyły) dorosłym i dzieciom. Jeśli droga dożylna (żyła lub rurka do żyły) nie jest dostępna, lekarz może zdecydować, czy wstrzyknąć EPREX pod skórę (podskórnie). W tym pacjentów dializowanych i pacjentów jeszcze nie dializowanych.
- Lekarz będzie miał regularne badania krwi, aby sprawdzić, czy niedokrwistość odpowiada i może dostosować dawkę, zwykle nie częściej niż raz na 4 tygodnie.
- Po ustąpieniu niedokrwistości lekarz będzie kontynuował regularne sprawdzanie wyników badań krwi Dawka leku EPREX i częstość podawania mogą być dalej dostosowywane w celu utrzymania odpowiedzi na leczenie.
- Jeśli pacjent jest leczony lekiem EPREX w dłuższych odstępach między dawkami (większymi niż raz w tygodniu), może nie być w stanie odpowiednio utrzymać poziomu hemoglobiny i może być konieczne zwiększenie dawki leku EPREX lub częstości jego podawania.
- Aby leczenie było skuteczniejsze, suplementy żelaza mogą być przydatne przed i w trakcie leczenia preparatem EPREX.
- Jeśli pacjent jest poddawany hemodializie na początku leczenia lekiem EPREX, może być konieczne dostosowanie schematu dializy. Lekarz zdecyduje.
Dorośli pacjenci poddawani chemioterapii
- Lekarz może rozpocząć leczenie lekiem EPREX, jeśli stężenie hemoglobiny wynosi 10 g/dl lub mniej.
- Twoja wartość hemoglobiny będzie utrzymywana między 10 a 12 g / dl, ponieważ wyższy poziom hemoglobiny może zwiększać ryzyko zakrzepicy i śmierci.
- Dawka początkowa wynosi 150 jm na kilogram masy ciała 3 razy w tygodniu lub 450 jm na kilogram masy ciała raz w tygodniu.
- EPREX podaje się we wstrzyknięciu podskórnym.
- Lekarz będzie miał regularne badania krwi i może dostosować dawkę w zależności od odpowiedzi pacjenta na leczenie lekiem EPREX.
- Aby leczenie było skuteczniejsze, przed i w trakcie leczenia preparatem EPREX może być przydatny suplement żelaza.
- Leczenie EPREX jest zwykle kontynuowane przez 1 miesiąc po zakończeniu chemioterapii.
Dorośli pacjenci, którzy deponują własną krew
- Zazwyczaj stosowana dawka to 600 jm na kilogram masy ciała 2 razy w tygodniu.
- EPREX podaje się dożylnie bezpośrednio po odłożeniu krwi przez 3 tygodnie przed zabiegiem.
- Aby leczenie było skuteczniejsze, przed i w trakcie leczenia preparatem EPREX może być przydatny suplement żelaza.
Dorośli pacjenci, którzy są kandydatami do poważnej operacji ortopedycznej
- Zalecana dawka to 600 jm na kilogram masy ciała raz w tygodniu.
- EPREX podaje się we wstrzyknięciu podskórnym co tydzień przez 3 tygodnie przed zabiegiem oraz w dniu zabiegu.
- W przypadku konieczności skrócenia czasu przed zabiegiem, dawka dobowa 300 j.m./kg zostanie podana w ciągu 10 dni przed zabiegiem, w dniu zabiegu iw ciągu 4 dni po zabiegu.
- Jeśli badania krwi przed operacją wykażą zbyt wysokie wartości hemoglobiny, leczenie zostanie przerwane.
- Aby leczenie było skuteczniejsze, suplementy żelaza mogą być przydatne przed i w trakcie leczenia preparatem EPREX.
Instrukcje samodzielnego wstrzykiwania EPREX
- Na początku leczenia EPREX jest zwykle podawany przez lekarza lub pielęgniarkę.Następnie lekarz może zasugerować, aby pacjent (lub jego opiekun) nauczył się wstrzykiwać (podskórnie).
- Nie należy podejmować prób samodzielnego wstrzyknięcia, jeśli lekarz lub pielęgniarka nie poinstruowali, jak to zrobić.
- Zawsze postępuj zgodnie z instrukcjami lekarza lub pielęgniarki.
- Stosować EPREX tylko wtedy, gdy był prawidłowo przechowywany - patrz rozdział Jak przechowywać EPREX
- Przed użyciem wyjmij strzykawkę Eprex z lodówki i pozwól jej osiągnąć temperaturę pokojową.Zazwyczaj zajmuje to 15-30 minut.
Pobrać pojedynczą dawkę EPREXu z każdej ampułko-strzykawki.Gdy EPREX jest podawany pod skórę (podskórnie), objętość zwykle nie przekracza 1 mililitra (1 ml) dla pojedynczego wstrzyknięcia.
EPREX należy podawać samodzielnie i nie mieszać z innymi płynami do wstrzykiwań.
Nie wstrząsać ampułko-strzykawkami EPREX. Długotrwałe energiczne potrząsanie może uszkodzić produkt. Nie używać produktu, jeśli został energicznie wstrząśnięty.
Jak samodzielnie wykonać wstrzyknięcie za pomocą ampułko-strzykawek
Ampułko-strzykawki są wyposażone w zabezpieczenie igły PROTECS™, zapobiegające ryzyku ukłucia się igłą po użyciu. Jest to wskazane na opakowaniu.
- Przed użyciem wyjąć ampułko-strzykawkę z lodówki.Płyn musi osiągnąć temperaturę pokojową.Nie zdejmować nasadki ochronnej z igły podczas oczekiwania, aż osiągnie temperaturę pokojową.
- Należy sprawdzić ampułko-strzykawkę, aby upewnić się, że jest to właściwa dawka, że nie upłynął jej termin ważności, czy nie jest uszkodzona, a płyn jest przezroczysty i niezamrożony.
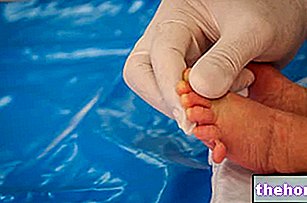
- Wybierz miejsce wstrzyknięcia.Najbardziej odpowiednie miejsca do wstrzyknięcia to udo i brzuch, z wyjątkiem okolicy pępka. Za każdym razem zmieniać miejsce wstrzyknięcia.
- Do mycia rąk. Użyj antyseptycznej chusteczki do dezynfekcji miejsca wstrzyknięcia.
- Trzymać ampułko-strzykawkę za cylinder strzykawki z zakrytą igłą skierowaną do góry.
- Nie trzymać za głowicę tłoka, tłok, skrzydełko osłony igły lub nasadkę osłony igły.
- Pod żadnym pozorem nie odciągaj tłoka
- Nie zdejmować nasadki z igły ampułko-strzykawki do czasu, gdy pacjent będzie gotowy do podania leku EPREX
- Zdjąć nasadkę ochronną z igły strzykawki, trzymając ją za korpus i pociągając nasadkę bez obracania. Nie naciskać tłoka, nie dotykać igły ani nie potrząsać strzykawką. - Nie dotykaj klipsów aktywujących urządzenie zabezpieczające, aby zapobiec przedwczesnemu zakryciu igły klapką zabezpieczającą igłę
- Unieś skórę między kciuk i palec wskazujący, nie ściskając jej zbyt mocno.
- Wbić igłę do końca. Lekarz lub pielęgniarka pokażą, jak to zrobić.
- Nacisnąć tłok kciukiem, aż zostanie wstrzyknięta pełna ilość płynu odpowiadająca prawidłowej dawce. Wciskać powoli i równomiernie, utrzymując skórę uniesioną. Urządzenie zabezpieczające igłę PROTECS™ nie uruchomi się, dopóki wstrzyknięcie nie zostanie wykonane. pełna dawka. Możesz usłyszeć „kliknięcie”, gdy aktywowane jest zabezpieczenie igły PROTECS ™.
- Gdy tłok osiągnie koniec skoku, wyciągnąć igłę i uwolnić skórę.
- Powoli zdejmij kciuk z tłoka, aby całkowicie zasłonić igłę przez urządzenie zabezpieczające.
- Pod koniec wstrzyknięcia w miejscu wstrzyknięcia może pojawić się niewielka ilość krwi. Jest to normalne. Miejsce wstrzyknięcia można zdezynfekować, naciskając nakładkę antyseptyczną na kilka sekund.
- Zużyte strzykawki należy wyrzucić do bezpiecznego pojemnika – patrz punkt Jak przechowywać lek Eprex
Przedawkowanie Co zrobić w przypadku przyjęcia zbyt dużej dawki leku Eprex
Zastosowanie większej niż zalecana dawki EPREX
Jeśli pacjent uważa, że zastosował zbyt dużą dawkę leku EPREX, powinien natychmiast powiedzieć o tym lekarzowi lub pielęgniarce.. Działania niepożądane wynikające z przedawkowania są mało prawdopodobne.
Pominięcie wstrzyknięcia leku EPREX
Kolejne wstrzyknięcie należy wykonać, gdy tylko sobie o tym przypomnimy.Jeśli następne wstrzyknięcie przypada w ciągu jednego dnia, należy pominąć pominiętą dawkę i postępować zgodnie ze zwykłym harmonogramem. Nie podwajaj zastrzyków.
Jeśli jesteś pacjentem z wirusowym zapaleniem wątroby typu C i otrzymujesz interferon i rybawirynę
Należy zgłosić się do lekarza, ponieważ połączenie epoetyny alfa z interferonem i rybawiryną w rzadkich przypadkach prowadziło do utraty działania i rozwoju ciężkiej postaci niedokrwistości zwanej aplazją czystoczerwonokrwinkową (PRCA). leczenie niedokrwistości związanej z wirusowym zapaleniem wątroby typu C.
W przypadku dalszych pytań dotyczących stosowania tego produktu należy zwrócić się do lekarza, pielęgniarki lub farmaceuty.
Skutki uboczne Jakie są skutki uboczne leku Eprex
Jak każdy lek, EPREX może powodować działania niepożądane, chociaż nie u każdego one wystąpią. Należy natychmiast poinformować lekarza lub pielęgniarkę, jeśli wystąpi którykolwiek z wymienionych poniżej objawów.
Bardzo częste działania niepożądane
Występują u więcej niż 1 na 10 pacjentów stosujących EPREX
- Biegunka
- Uczucie mdłości w żołądku
- On wymiotował
- Gorączka
- Przekrwienie dróg oddechowych, takie jak zatkany nos i ból gardła, zgłaszano u pacjentów z chorobą nerek niepoddawanych jeszcze dializie.
Częste skutki uboczne
Dotyczą one do 1 na 10 pacjentów stosujących EPREX
- Podnoszenie wartości ciśnienia krwi. Ból głowy (zwłaszcza nagły, ostry i przypominający migrenę) lub splątanie lub drgawki. Mogą to być objawy nagłego wzrostu ciśnienia krwi, który wymaga natychmiastowego leczenia. Wzrost ciśnienia krwi może wymagać leczenia. dostosowanie dawki leków, które już przyjmujesz na nadciśnienie).
- Zakrzepy krwi (w tym zakrzepica żył głębokich i zator), które mogą wymagać pilnego leczenia. Objawy mogą obejmować ból w klatce piersiowej, świszczący oddech, bolesny obrzęk kończyn dolnych i zaczerwienienie, zwykle nóg.
- kaszel.
- Podrażnienie skóry, które może być spowodowane reakcją alergiczną.
- Ból kości lub mięśni.
- Zespół grypopodobny, taki jak ból głowy, bóle stawów, uczucie osłabienia, dreszcze, zmęczenie i zawroty głowy. Reakcje te są częstsze na początku leczenia. Jeśli wystąpią takie objawy podczas wstrzyknięcia dożylnego, wolniejsze podawanie może pomóc w ich uniknięciu w przyszłości.
- Zaczerwienienie, pieczenie i ból w miejscu wstrzyknięcia.
- Obrzęk kostek, stóp lub palców.
Niezbyt częste skutki uboczne
Dotyczą one do 1 na 100 pacjentów stosujących EPREX
- Wysokie stężenie potasu we krwi, które może powodować nieprawidłowy rytm serca (jest to bardzo częste działanie niepożądane u pacjentów dializowanych).
- Drgawki
- Zatkany nos lub drogi oddechowe
Bardzo rzadkie skutki uboczne
Dotyczą one do 1 na 10 000 pacjentów stosujących EPREX
- Objawy aplazji czysto czerwonych krwinek (PRCA). PRCA oznacza niezdolność do wytworzenia wystarczającej ilości czerwonych krwinek w szpiku kostnym. PRCA może powodować nagłą i ciężką anemię, której objawami są:
- niezwykłe zmęczenie,
- uczucie zawrotów głowy,
- duszność.
PRCA stwierdzano bardzo rzadko, szczególnie u pacjentów z chorobą nerek po miesiącach lub latach leczenia preparatem EPREX i innymi substancjami stymulującymi wytwarzanie krwinek czerwonych. Może wystąpić zwiększenie liczby małych krwinek (zwanych płytkami krwi), zwykle biorących udział w tworzeniu skrzepów, zwanych płytkami krwi, szczególnie na początku leczenia. Lekarz to sprawdzi.
Jeśli jesteś na hemodializie:
- W zespoleniu dializacyjnym mogą tworzyć się skrzepy krwi (zakrzepica). Może się to zdarzyć łatwiej, jeśli masz niskie ciśnienie krwi (niedociśnienie) lub jeśli masz problemy z przetoką.
- W systemie hemodializy mogą również tworzyć się skrzepy krwi.Lekarz może zdecydować o zwiększeniu dawki heparyny podczas dializy.
Należy natychmiast powiedzieć lekarzowi lub pielęgniarce, jeśli pacjent zauważy którykolwiek z tych objawów lub jeśli podczas przyjmowania leku EPREX wystąpią jakiekolwiek inne objawy.
Jeśli którykolwiek z objawów niepożądanych nasili się lub wystąpią jakiekolwiek objawy niepożądane niewymienione w tej ulotce, należy poinformować o tym lekarza, pielęgniarkę lub farmaceutę.
Dla osób uprawiających sport: stosowanie leku bez konieczności terapeutycznej stanowi doping i może w każdym przypadku decydować o pozytywnych wynikach testów antydopingowych.
Wygaśnięcie i przechowywanie
Przechowywać w miejscu niewidocznym i niedostępnym dla dzieci.
Nie stosować tego leku po upływie terminu ważności zamieszczonego na pudełku i etykiecie po literach EXP.Termin ważności przypada na ostatni dzień wskazanego miesiąca.
EPREX należy przechowywać w lodówce (2°C - 8°C).
EPREX można wyjąć z lodówki i przechowywać w temperaturze pokojowej (nie wyższej niż 25°C) do 3 dni. Po wyjęciu ampułko-strzykawki z lodówki i osiągnięciu temperatury pokojowej (nie wyższej niż 25°C) należy ją zużyć w ciągu 3 dni lub wyrzucić.
Nie wolno go zamrażać ani wstrząsać
Przechowywać w oryginalnym opakowaniu, aby chronić je przed światłem.
Nie stosować tego leku, jeśli plomba jest uszkodzona lub jeśli roztwór jest zabarwiony lub widoczne są zawieszone cząstki. Jeśli te warunki są przestrzegane, wyrzuć lek
Leków nie należy wyrzucać do kanalizacji ani domowych pojemników na odpadki. Należy zapytać farmaceutę, jak usunąć leki, których się już nie używa, co pomoże chronić środowisko.
Zawartość opakowania i inne informacje
Co zawiera lek Eprex
Substancją czynną jest: Epoetyna alfa (ilość, patrz poniższa tabela).
Pozostałe składniki to: polisorbat 80, chlorek sodu, sodu wodorofosforan jednowodny, sodu fosforan dwuwodny, glicyna i woda do wstrzykiwań. Ten lek zawiera mniej niż 1 mmol sodu (23 mg) na dawkę, więc zasadniczo nie zawiera sodu.
Jak wygląda lek EPREX i co zawiera opakowanie
EPREX to roztwór do wstrzykiwań w ampułko-strzykawkach. Ampułko-strzykawki są wyposażone w zabezpieczenie igły PROTECS™ (patrz tabela poniżej) EPREX jest przezroczystym i bezbarwnym roztworem.
Nie wszystkie rozmiary opakowań mogą być wprowadzone na rynek
Ulotka pakietu źródłowego: AIFA (Włoska Agencja Leków). Treść opublikowana w styczniu 2016 r. Przedstawione informacje mogą być nieaktualne.
Aby mieć dostęp do najbardziej aktualnej wersji, warto wejść na stronę AIFA (Włoskiej Agencji Leków). Zastrzeżenie i przydatne informacje.
01.0 NAZWA PRODUKTU LECZNICZEGO
EPREX 10000 IU / ML ROZTWÓR DO WSTRZYKIWAŃ W AMPUŁKO-STRZYKAWCE
02.0 SKŁAD JAKOŚCIOWY I ILOŚCIOWY
Epoetyna alfa 10 000 j.m./ml (84,0 mcg na ml) wytworzona technologią rekombinacji DNA w komórkach jajnika chomika chińskiego (CHO).
Jedna ampułko-strzykawka 0,3 ml zawiera 3000 j.m. (25,2 mcg) epoetyny alfa
Jedna ampułko-strzykawka o pojemności 0,4 ml zawiera 4000 j.m. (33,6 mcg) epoetyny alfa
Jedna ampułko-strzykawka 0,5 ml zawiera 5000 j.m. (42,0 mcg) epoetyny alfa
Jedna ampułko-strzykawka 0,6 ml zawiera 6000 j.m. (50,4 mikrograma) epoetyny alfa
Jedna ampułko-strzykawka 0,8 ml zawiera 8000 j.m. (67,2 mcg) epoetyny alfa
Jedna ampułko-strzykawka o pojemności 1,0 ml zawiera 10 000 j.m. (84,0 mcg) epoetyny alfa
Ten produkt leczniczy zawiera mniej niż 1 mmol (23 mg) sodu na dawkę, co oznacza, że zasadniczo jest „wolny od sodu”.
Pełny wykaz substancji pomocniczych, patrz punkt 6.1
03.0 POSTAĆ FARMACEUTYCZNA
Roztwór do wstrzykiwań w ampułko-strzykawkach
Przezroczysty, bezbarwny roztwór
04.0 INFORMACJE KLINICZNE
04.1 Wskazania terapeutyczne
EPREX jest wskazany w leczeniu objawowej niedokrwistości związanej z przewlekłą niewydolnością nerek (CRI):
• u dorosłych i dzieci w wieku od 1 do 18 lat poddawanych hemodializie oraz u dorosłych pacjentów poddawanych dializie otrzewnowej.
• u dorosłych pacjentów z niewydolnością nerek niepoddawanych jeszcze dializie w leczeniu ciężkiej niedokrwistości pochodzenia nerkowego z towarzyszącymi objawami klinicznymi u pacjentów.
EPREX jest wskazany do stosowania u dorosłych pacjentów otrzymujących chemioterapię z powodu guzów litych, chłoniaka złośliwego lub szpiczaka mnogiego, u których występuje ryzyko transfuzji, zgodnie z ogólnym stanem pacjenta (sytuacja sercowo-naczyniowa, istniejąca niedokrwistość na początku chemioterapii) w leczeniu niedokrwistości i redukcji potrzeby transfuzji.
EPREX jest wskazany u pacjentów dorosłych, którzy biorą udział w programie predoncji w celu zwiększenia ilości krwi autologicznej.Leczenie wskazane jest tylko u pacjentów z umiarkowaną niedokrwistością (stężenie hemoglobiny w zakresie Hb 10-13 g/dl [6 , 2 – 8,1 mmol/l], brak niedoboru żelaza), jeśli procedury przechowywania krwi nie są dostępne lub niewystarczające w przypadku dużego zabiegu chirurgicznego wymagającego dużej ilości krwi (4 lub więcej jednostek na kobietę lub 5 lub więcej jednostek dla mężczyzn).
EPREX jest wskazany u dorosłych pacjentów, u których nie występuje niedobór żelaza, przed dużym planowym zabiegiem ortopedycznym, u których uważa się, że istnieje wysokie ryzyko powikłań po przetoczeniu, w celu zmniejszenia narażenia na przetoczenia krwi allogenicznej. stężenie hemoglobiny w zakresie Hb10-13 g/dl), u których nie jest dostępny program autologicznego przetoczenia krwi i u których przewiduje się umiarkowaną utratę krwi (od 900 do 1800 ml).
04.2 Dawkowanie i sposób podawania
Dawkowanie
Przed rozpoczęciem leczenia epoetyną alfa i przy podejmowaniu decyzji o zwiększeniu dawki wszystkie inne przyczyny niedokrwistości (niedobór żelaza, kwasu foliowego lub witaminy B12, zatrucie glinem, zakażenia lub stany zapalne, utrata krwi, hemoliza i zwłóknienie szpiku kostnego dowolnego pochodzenia). W celu zapewnienia optymalnej odpowiedzi na epoetynę alfa należy zapewnić odpowiednie zapasy żelaza i w razie potrzeby zastosować suplementację żelaza (patrz punkt 4.4).
Leczenie objawowej niedokrwistości u dorosłych pacjentów z przewlekłą niewydolnością nerek (CRI)
Objawy i następstwa niedokrwistości mogą się różnić w zależności od płci, wieku i współistniejących chorób; konieczna jest ocena stanu klinicznego danego pacjenta przez lekarza.
Pożądane stężenie hemoglobiny wynosi od 10 g/dl do 12 g/dl (6,2 do 7,5 mmol/l). EPREX należy podawać w taki sposób, aby osiągnąć stężenie hemoglobiny nie przekraczające 12 g/dl (7,5 mmol/l). Należy unikać wzrostu stężenia hemoglobiny o ponad 2 g/dl (1,25 mmol/l) w okresie czterech tygodni. W takim przypadku należy odpowiednio dostosować dawkę.
Ze względu na zmienność wewnątrzosobniczą u pacjenta mogą czasami wystąpić wartości hemoglobiny powyżej i poniżej pożądanego stężenia hemoglobiny. Tę zmienność należy kontrolować poprzez dostosowanie dawki, uwzględniając zakres stężenia hemoglobiny od 10 g/dl (6,2 mmol/l) do 12 g/dl (7,5 mmol/l).
Należy unikać stałego poziomu hemoglobiny powyżej 12 g/dl (7,5 mmol). Jeśli stężenie hemoglobiny wzrasta o więcej niż 2 g/dL (1,25 mmol/L) miesięcznie lub jeśli poziom hemoglobiny stale przekracza 12 g/dL (7,5 mmol), zmniejsz dawkę EPREX o 25%.Jeżeli stężenie hemoglobiny przekracza 13 g/dl (8,1 mmol/l), wstrzymaj terapię do czasu powrotu do poziomu 12 g/dl (7,5 mmol/l), a następnie kontynuuj EPREX w dawkach o 25% niższych niż poprzednie.
Pacjentów należy ściśle monitorować, aby upewnić się, że stosowana jest najniższa dopuszczona skuteczna dawka preparatu EPREX w celu zapewnienia odpowiedniej kontroli niedokrwistości i powiązanych objawów poprzez utrzymanie stężenia hemoglobiny poniżej lub równego 12 g/dl (7, 5 mmol/l).
Należy zachować ostrożność przy zwiększaniu dawki ESA u pacjentów z przewlekłą niewydolnością nerek.W przypadku pacjentów ze słabą odpowiedzią hemoglobiny na ESA należy szukać alternatywnej przyczyny słabej odpowiedzi (patrz punkty 4.4 i 5.1).
Zabieg EPREX dzieli się na dwie fazy - fazę korekcji i fazę podtrzymującą.
Dorośli pacjenci poddawani hemodializie
U pacjentów poddawanych hemodializie, u których dostęp żylny jest łatwo dostępny, preferowane jest podawanie drogą dożylną.
Faza korekty :
Dawka początkowa to 50 jm/kg masy ciała 3 razy w tygodniu.
W razie potrzeby zwiększ lub zmniejsz dawkę o 25 j.m./kg (3 razy w tygodniu) aż do uzyskania pożądanego stężenia hemoglobiny w zakresie od 10g/dl do 12g/dl (6,2 do 7,5 mmol/l) (należy to robić stopniowo o odstępach co najmniej cztery tygodnie).
- Faza konserwacji :
Zalecana całkowita dawka tygodniowa wynosi od 75 j.m./kg do 300 j.m./kg.
Należy odpowiednio dostosować dawkę, aby utrzymać wartości hemoglobiny w pożądanym stężeniu hemoglobiny między 10 g/dl a 12 g/dl (6,2 do 7,5 mmol/l).
Pacjenci z bardzo niskim początkowym poziomem hemoglobiny (8 g/dl lub > 5 mmol/l).
Dorośli pacjenci z niewydolnością nerek niepoddani jeszcze dializie
U pacjentów, u których dostęp żylny nie jest łatwo dostępny, EPREX można podawać podskórnie.
Faza korekty
Dawka początkowa wynosi 50 j.m./kg 3 razy w tygodniu, a następnie, jeśli to konieczne, zwiększaj dawkę o 25 j.m./kg (3 razy w tygodniu) aż do osiągnięcia pożądanego stężenia hemoglobiny (należy to robić stopniowo. odstępach co najmniej cztery tygodnie).
Faza konserwacji
W fazie podtrzymującej EPREX można podawać 3 razy w tygodniu, a w przypadku podawania podskórnego raz w tygodniu lub raz na dwa tygodnie.
Aby utrzymać wartości hemoglobiny na pożądanym poziomie, należy odpowiednio dostosować dawki i odstępy między dawkami: Hb między 10 a 12 g/dl (6,2-7,5 mmol/l). Wydłużenie odstępu między dawkami może wymagać zwiększenia dawki.
Maksymalna dawka nie powinna przekraczać 150 jm/kg 3 razy w tygodniu, 240 jm/kg (maksymalnie 20 000 jm) raz w tygodniu lub 480 jm/kg (maksymalnie 40 000 jm) raz na 2 tygodnie.
Dorośli pacjenci poddawani dializie otrzewnowej
U pacjentów, u których dostęp żylny nie jest łatwo dostępny, EPREX można podawać podskórnie.
Faza korekty
Dawka początkowa to 50 jm/kg, dwa razy w tygodniu.
Faza konserwacji
Zalecana dawka podtrzymująca wynosi od 25 j.m./kg do 50 j.m./kg dwa razy w tygodniu, podzielona na 2 równe podania.
Należy odpowiednio dostosować dawkę, aby utrzymać wartości hemoglobiny na pożądanym poziomie: hemoglobina między 10 g/dl a 12 g/dl (6,2-7,5 mmol/l).
Leczenie dorosłych pacjentów z niedokrwistością wywołaną chemioterapią
Objawy i następstwa anemii mogą się różnić w zależności od wieku, płci i ogólnej sytuacji choroby; konieczna jest ocena stanu klinicznego danego pacjenta przez lekarza.
EPREX należy podawać pacjentom z niedokrwistością, np. stężenie hemoglobiny ≤10 g/dl (6,2 mmol/l).
Dawka początkowa wynosi 150 jm/kg, podawana podskórnie 3 razy w tygodniu.
Alternatywnie, EPREX można podawać podskórnie w dawce początkowej 450 jm/kg raz w tygodniu.
Należy odpowiednio dostosować dawkę, aby utrzymać wartości hemoglobiny na pożądanym poziomie: hemoglobina między 10 g/dl a 12 g/dl (6,2-7,5 mmol/l).
Ze względu na zmienność wewnątrzosobniczą u pacjenta mogą czasami wystąpić wartości hemoglobiny powyżej i poniżej pożądanego stężenia hemoglobiny. Tę zmienność należy kontrolować poprzez dostosowanie dawki, uwzględniając pożądany zakres stężenia hemoglobiny od 10 g/dl (6,2 mmol/l) do 12 g/dl (7,5 mmol/l).
Należy unikać stałego stężenia hemoglobiny powyżej 12 g/dl (7,5 mmol). Instrukcje dotyczące prawidłowego dostosowania dawki w przypadku osiągnięcia przez hemoglobinę stężenia powyżej 12 g/dl (7,5 mmol) znajdują się poniżej.
Jeżeli po 4 tygodniach leczenia stężenie hemoglobiny wzrosło o co najmniej 1 g/dl (0,62 mmol/l) lub liczba retikulocytów wzrosła o ≥ 40 000 komórek/mcl w stosunku do wartości wyjściowej, dawka powinna pozostać na poziomie 150 j.m./ kg 3 razy w tygodniu lub 450 IU / kg raz w tygodniu.
Jeśli wzrost stężenia hemoglobiny wynosi
Jeśli wzrost stężenia hemoglobiny był
Dostosowanie dawki w celu utrzymania stężenia hemoglobiny w granicach 10 g/dl – 12 g/dl
Jeśli stężenie hemoglobiny wzrośnie o więcej niż 2 g/dl (1,25 mmol/l) miesięcznie lub jeśli hemoglobina przekroczy 12 g/dl (7,5 mmol/l), zmniejsz dawkę EPREX o około 25 -50%.
Jeżeli stężenie hemoglobiny przekroczy 13 g/dl (8,1 mmol/l), należy przerwać terapię do momentu, gdy stężenie spadnie poniżej 12 g/dl (7,5 mmol/l), a następnie wznowić terapię preparatem EPREX w dawce 25% niższej niż poprzednia dawka.
Zalecane dawkowanie opisane jest na poniższym schemacie:
Pacjentów należy ściśle monitorować, aby upewnić się, że do odpowiedniej kontroli objawów niedokrwistości stosowana jest najniższa zatwierdzona dawka środków stymulujących erytropoezę (ESA).
Terapię EPREX należy kontynuować przez miesiąc po zakończeniu chemioterapii.
Leczenie dorosłych pacjentów, którzy są kandydatami do zabiegu chirurgicznego w ramach programu autologicznego przetoczenia krwi
Pacjenci z łagodną niedokrwistością (hematokryt 33-39%), wymagający wstępnego odłożenia 4 lub więcej jednostek krwi, powinni być leczeni dożylnie 600 jm/kg EPREXu, dwa razy w tygodniu, przez 3 tygodnie przed zabiegiem.
EPREX należy podawać po zakończeniu procedury oddawania krwi.
Leczenie dorosłych pacjentów będących kandydatami do dużych planowych operacji ortopedycznych
Zalecana dawka to 600 jm/kg EPREXu podawanego podskórnie, raz w tygodniu przez 3 tygodnie przed zabiegiem (-21 dni, -14 dni i -7 dni) oraz w dniu zabiegu.
Jeżeli istnieje medyczna potrzeba skrócenia czasu oczekiwania przed zabiegiem do mniej niż 3 tygodni, dawkę 300 j.m./kg EPREXu należy podawać codziennie podskórnie przez 10 kolejnych dni przed zabiegiem, w dniu zabiegu oraz w 4 dni bezpośrednio po nim.
Jeśli stężenie hemoglobiny osiągnie 15 g/dl lub więcej w okresie przedoperacyjnym, podawanie produktu EPREX należy przerwać i nie należy podawać kolejnych dawek.
Populacja pediatryczna
Leczenie objawowej niedokrwistości u pacjentów z przewlekłą niewydolnością nerek poddawanych hemodializie
Objawy i następstwa niedokrwistości mogą się różnić w zależności od wieku, płci i współistniejących chorób; konieczna jest ocena stanu klinicznego danego pacjenta przez lekarza.
U pacjentów pediatrycznych pożądane stężenie hemoglobiny wynosi od 9,5 g/dl do 11 g/dl (5,9 do 6,8 mmol/l). EPREX należy podawać w taki sposób, aby osiągnąć stężenie hemoglobiny nie przekraczające 11 g/dl (6,8 mmol/l). Należy unikać wzrostu stężenia hemoglobiny o ponad 2 g/dl (1,25 mmol/l) w okresie czterech tygodni. W takim przypadku należy odpowiednio dostosować dawkę.
Pacjenci powinni być ściśle monitorowani, aby upewnić się, że stosowana jest najniższa zatwierdzona dawka preparatu EPREX w celu zapewnienia odpowiedniej kontroli niedokrwistości i powiązanych objawów.
Zabieg EPREX dzieli się na dwie fazy: fazę korekcji i fazę podtrzymującą.
U pacjentów pediatrycznych poddawanych hemodializie, u których dostęp dożylny jest już dostępny, preferowane jest podawanie dożylne.
Faza korekty:
Dawka początkowa wynosi 50 jm / kg wagi dożylnie, 3 razy w tygodniu.
W razie potrzeby zwiększ lub zmniejsz dawkę o 25 j.m./kg (3 razy w tygodniu) aż do uzyskania pożądanego stężenia hemoglobiny w zakresie od 9,5 g/dl do 11 g/dl (5,9 do 6, 8 mmol/l) (powinno to nastąpić stopniowo w odstępach co najmniej czterech tygodni).
Faza konserwacji :
Należy odpowiednio dostosować dawkę, aby utrzymać wartości hemoglobiny w pożądanym stężeniu hemoglobiny między 9,5 g/dl a 11 g/dl (5,9 do 6,8 mmol/l).
Ogólnie rzecz biorąc, dzieci o masie ciała poniżej 30 kg wymagają wyższych dawek podtrzymujących niż dzieci o masie ciała powyżej 30 kg i dorośli.
Pacjenci pediatryczni z bardzo niskim początkowym poziomem hemoglobiny (6,8 g/dL lub >4,25 mmol/L).
Leczenie pacjentów pediatrycznych z niedokrwistością wywołaną chemioterapią.
Nie ustalono bezpieczeństwa i skuteczności EPREX u dzieci i młodzieży otrzymujących chemioterapię.
Leczenie pediatrycznych pacjentów chirurgicznych uczestniczących w programie autologicznej predoncji
Bezpieczeństwo i skuteczność preparatu EPREX u dzieci nie zostały ustalone.
Brak danych.
Leczenie pacjentów pediatrycznych oczekujących na duży planowy zabieg ortopedyczny
Bezpieczeństwo i skuteczność preparatu EPREX u dzieci nie zostały ustalone.
Brak danych.
Sposób podawania
Zachowaj ostrożność przed użyciem lub podaniem leku.
Przed użyciem odstawić strzykawkę EPREX do temperatury pokojowej, co zwykle zajmuje od 15 do 30 minut.
Leczenie objawowej niedokrwistości u dorosłych pacjentów z przewlekłą niewydolnością nerek
U pacjentów z przewlekłą niewydolnością nerek, u których dostęp dożylny jest normalnie dostępny (pacjenci poddawani hemodializie), preferowane jest podawanie dożylne preparatu EPREX.
Jeżeli dostęp dożylny nie jest łatwo dostępny (pacjenci jeszcze nie poddawani dializie i pacjenci poddawani dializie otrzewnowej), EPREX można podawać podskórnie.
Leczenie dorosłych pacjentów z niedokrwistością wywołaną chemioterapią.
EPREX należy podawać podskórnie.
Leczenie dorosłych pacjentów chirurgicznych uczestniczących w programie autologicznej predoncji
EPREX należy podawać dożylnie.
Leczenie dorosłych pacjentów zakwalifikowanych do poważnej planowej operacji ortopedycznej
EPREX należy podawać podskórnie.
Leczenie objawowej niedokrwistości u dzieci i młodzieży z przewlekłą niewydolnością nerek poddawanych hemodializie
U dzieci i młodzieży z przewlekłą niewydolnością nerek, u których dostęp dożylny jest już dostępny (pacjenci poddawani hemodializie), preferowane jest podawanie dożylne preparatu EPREX.
Podawanie dożylne
Podawanie powinno zająć co najmniej 1-5 minut w zależności od całkowitej dawki.
U pacjentów poddawanych hemodializie bolus można podać podczas dializy przez odpowiedni dostęp żylny w linii dializacyjnej lub wstrzyknięcie można podać pod koniec dializy przez dostęp do przetoki, a następnie podać 10 ml roztworu fizjologicznego do płukania dróg dostępu i zapewnienia zadowalającego wprowadzenia produktu do krwiobiegu.
U pacjentów, u których wystąpiły reakcje grypopodobne, preferowane jest wolniejsze podawanie (patrz punkt 4.8).
Nie podawać EPREXu we wlewie dożylnym lub w roztworze z innymi lekami.
Podanie podskórne
Zasadniczo nie należy przekraczać maksymalnej objętości 1 ml w każdym miejscu wstrzyknięcia. W przypadku większych objętości należy wybrać więcej niż jedno miejsce wstrzyknięcia.
Zastrzyki należy wykonywać w kończyny lub przednią ścianę brzucha.
W przypadku, gdy lekarz uzna, że pacjent lub opiekun jest w stanie bezpiecznie i prawidłowo podać EPREX podskórnie, należy dostarczyć instrukcje dotyczące właściwej dawki i podawania.
Podobnie jak w przypadku innych produktów do wstrzykiwania, należy sprawdzić, czy w roztworze nie ma cząstek ani zmian kolorystycznych.
04.3 Przeciwwskazania
Nadwrażliwość na substancję czynną lub na którąkolwiek substancję pomocniczą.
Pacjenci, u których wystąpiła wybiórcza aplazja czerwonokrwinkowa (PRCA) po leczeniu jakąkolwiek erytropoetyną, nie powinni być leczeni preparatem EPREX lub innymi erytropoetynami (patrz punkt 4.4 PRCA).
Niekontrolowane nadciśnienie.
U pacjentów leczonych preparatem EPREX należy pamiętać o wszystkich przeciwwskazaniach związanych z programem wstępnego osadzania krwi autologicznej.
Stosowanie preparatu EPREX jest przeciwwskazane w przypadku wystąpienia ciężkich zaburzeń naczyniowych na poziomie tętnic wieńcowych, obwodowych, tętnic szyjnych lub mózgowych u pacjentów, którzy są kandydatami do dużych planowych operacji ortopedycznych i nie są częścią programu autologicznego przetoczenia. u pacjentów z niedawno przebytymi epizodami zawału mięśnia sercowego lub innymi powikłaniami naczyniowo-mózgowymi.
Pacjenci będący kandydatami do operacji, którzy z jakiegokolwiek powodu nie mogą otrzymać odpowiedniej profilaktyki przeciwzakrzepowej.
04.4 Specjalne ostrzeżenia i odpowiednie środki ostrożności dotyczące stosowania
Ogólny
U wszystkich pacjentów otrzymujących epoetynę alfa ciśnienie krwi należy ściśle monitorować i w razie potrzeby kontrolować. Epoetynę alfa należy stosować ostrożnie w przypadku nieleczonego, niewłaściwie leczonego lub trudnego do kontrolowania nadciśnienia. Konieczne może być rozpoczęcie lub zintensyfikowanie leczenia przeciwnadciśnieniowego. Jeśli nie można kontrolować ciśnienia krwi, leczenie epoetyną alfa należy przerwać.
Kryzysy nadciśnieniowe z encefalopatią i drgawkami, wymagające natychmiastowej pomocy lekarskiej i intensywnej opieki medycznej, występowały podczas leczenia epoetyną alfa nawet u pacjentów z poprzednio prawidłowym lub niskim ciśnieniem krwi. Szczególną uwagę należy zwrócić na ukłucia przypominające migrenę jako możliwy znak ostrzegawczy (patrz punkt 4.8).
Epoetynę alfa należy stosować ostrożnie u pacjentów z padaczką, napadami drgawkowymi w wywiadzie lub stanami chorobowymi związanymi z predyspozycją do napadów drgawkowych, takimi jak zakażenia ośrodkowego układu nerwowego i przerzuty do mózgu.
Epoetynę alfa należy stosować ostrożnie u pacjentów z przewlekłą niewydolnością wątroby.Bezpieczeństwo stosowania epoetyny alfa u pacjentów z zaburzeniami czynności wątroby nie zostało ustalone.
U pacjentów, którzy otrzymali ESA (patrz punkt 4.8), obserwowano zwiększoną częstość występowania zakrzepicy naczyń (ŻChZZ), w tym zakrzepicy i zatoru żył i tętnic (w tym niektórych zakończonych zgonem), takich jak zakrzepica żył głębokich, zator tętnicy płucnej, zakrzepica i zawał mięśnia sercowego Ponadto zgłaszano incydenty naczyniowo-mózgowe (w tym zawał mózgu, krwotok mózgowy i przemijające napady niedokrwienne).
Ryzyko wystąpienia tych ŻChZZ należy starannie rozważyć w stosunku do korzyści z leczenia epoetyną alfa, szczególnie u pacjentów z istniejącymi wcześniej czynnikami ryzyka ŻChZZ, w tym otyłością i przebytą ŻChZZ (np. zakrzepicą żył głębokich, zatorowością płucną i incydentami naczyniowymi mózgu).
U wszystkich pacjentów należy ściśle monitorować stężenie hemoglobiny ze względu na potencjalne zwiększone ryzyko zdarzeń zakrzepowo-zatorowych i zgonów, gdy pacjenci są leczeni stężeniami hemoglobiny powyżej wskazanego stężenia.
Podczas leczenia epoetyną alfa może wystąpić umiarkowany, zależny od dawki wzrost liczby płytek krwi, ale w normalnym zakresie. Zjawisko to ustępuje w trakcie terapii. Ponadto zgłaszano nadpłytkowość powyżej normy Zaleca się regularne monitorowanie płytek krwi w ciągu pierwszych 8 tygodni leczenia.
Wszystkie możliwe przyczyny niedokrwistości (niedobór żelaza, niedobór kwasu foliowego lub witaminy B12, zatrucie glinem, zakażenie lub stan zapalny, utrata krwi, hemoliza i zwłóknienie szpiku kostnego dowolnego pochodzenia) należy ocenić i leczyć przed rozpoczęciem leczenia epoetyną alfa i podjęciem decyzji ma na celu zwiększenie dawki. W większości przypadków wartości ferrytyny w surowicy spadają wraz ze wzrostem wartości hematokrytu.W celu zapewnienia optymalnej odpowiedzi na epoetynę alfa należy zapewnić odpowiednie zapasy żelaza i w razie potrzeby podać suplementy żelaza ( patrz punkt 4.2):
• U pacjentów z przewlekłą niewydolnością nerek zaleca się suplementację żelaza, jeśli poziom ferrytyny jest niższy niż 100 ng/ml (żelazo pierwiastkowe dla dorosłych 200 do 300 mg/dobę doustnie i dla dzieci od 100 do 200 mg/dobę doustnie).
• W przypadku pacjentów z rakiem zaleca się suplementację żelaza, jeśli wysycenie transferyny wynosi poniżej 20% (żelazo pierwiastkowe 200 do 300 mg / dzień doustnie).
• W przypadku pacjentów w programie dawstwa autologicznego, suplementację żelaza (żelazo pierwiastkowe 200 mg/dobę doustnie) należy podawać na kilka tygodni przed rozpoczęciem dawstwa autologicznego w celu osiągnięcia wysokich zapasów żelaza. Przed rozpoczęciem leczenia epoetyną alfa i w trakcie kursu terapii epoetyną alfa.
• W przypadku pacjentów zakwalifikowanych do dużych planowych operacji ortopedycznych, suplementacja żelaza (żelazo pierwiastkowe 200 mg/dobę doustnie) powinna być podawana podczas leczenia epoetyną alfa. „suplementacja żelaza przed rozpoczęciem leczenia epoetyną alfa w celu osiągnięcia odpowiedniej rezerwy żelaza.
U pacjentów leczonych epoetyną alfa bardzo rzadko obserwowano początek lub zaostrzenie porfirii.
Epoetynę alfa należy stosować ostrożnie u pacjentów z porfirią.
W celu zapewnienia identyfikowalności środków stymulujących erytropoezę (ESA), nazwa handlowa podawanego ESA musi być zawsze zarejestrowana lub wskazana w dokumentacji medycznej pacjenta.
Zmiana terapii z jednego ESA na inny powinna odbywać się tylko pod odpowiednim nadzorem.
Aplazja czystej krwinek czerwonych (PRCA)
Aplazja wybiórczo czerwonych krwinek (PRCA) zależna od przeciwciał była zgłaszana po miesiącach lub latach, głównie u pacjentów z przewlekłą niewydolnością nerek leczonych epoetyną podawaną podskórnie.
Zgłaszano również przypadki u pacjentów z wirusowym zapaleniem wątroby typu C leczonych interferonem i rybawiryną w skojarzeniu z ESA. Epoetyna alfa nie jest zatwierdzona do leczenia niedokrwistości związanej z wirusowym zapaleniem wątroby typu C.
U pacjentów, u których występuje nagła utrata skuteczności, definiowana jako spadek wartości hemoglobiny (1 do 2 g/dl na miesiąc) ze zwiększoną potrzebą transfuzji, należy wykonać liczbę retikulocytów i ocenić znane przyczyny. odpowiedzi (takich jak niedobór żelaza, kwasu foliowego i witaminy B12, zatrucie glinem, infekcja lub stan zapalny, utrata krwi, hemoliza i zwłóknienie szpiku kostnego dowolnego pochodzenia).
Nieproporcjonalne zmniejszenie wartości hemoglobiny i rozwój ciężkiej niedokrwistości związanej z małą liczbą retikulocytów powinny prowadzić do przerwania leczenia epoetyną alfa i wykonania testu na obecność przeciwciał przeciwko erytropoetynie.
Należy również rozważyć badanie szpiku kostnego w celu rozpoznania PRCA.
Nie należy rozpoczynać leczenia innymi środkami ESA ze względu na ryzyko reakcji krzyżowej.
Leczenie objawowej niedokrwistości u dorosłych i dzieci z przewlekłą niewydolnością nerek (CRI):
U pacjentów z przewlekłą niewydolnością nerek otrzymujących epoetynę alfa należy regularnie mierzyć poziom hemoglobiny aż do osiągnięcia stabilnego poziomu, a następnie wykonywać jego pomiary okresowe.
U pacjentów z przewlekłą niewydolnością nerek w celu zmniejszenia ryzyka wzrostu ciśnienia krwi stężenie hemoglobiny powinno wzrosnąć o około 1 g/dl/miesiąc (0,62 mmol/l) i nie powinno przekraczać 2 g/dl/miesiąc (1,25 mmol/l).
U pacjentów z przewlekłą niewydolnością nerek podtrzymujące stężenie hemoglobiny nie powinno przekraczać maksymalnej wartości zakresu stężenia hemoglobiny podanej w punkcie 4.2 W badaniach klinicznych, w których stosowano środki stymulujące erytropoezę (ESA), obserwowano zwiększone ryzyko zgonu i poważnych zdarzeń sercowo-naczyniowych. podawane w celu osiągnięcia stężenia hemoglobiny powyżej 12 g/dl (7,5 mmol/ml).
Kontrolowane badania kliniczne nie wykazały znaczących korzyści związanych ze stosowaniem epoetyn, gdy stężenie hemoglobiny przekracza poziom niezbędny do opanowania objawów niedokrwistości i uniknięcia transfuzji krwi.
Należy zachować ostrożność przy zwiększaniu dawki preparatu EPREX u pacjentów z przewlekłą niewydolnością nerek, ponieważ duże skumulowane dawki epoetyny mogą wiązać się ze zwiększonym ryzykiem zgonu, poważnych zdarzeń sercowo-naczyniowych i mózgowo-naczyniowych. należy szukać przyczyn tej słabej reakcji (patrz sekcje 4.2 i 5.1).
Pacjenci z przewlekłą niewydolnością nerek leczeni podskórnie epoetyną alfa powinni być regularnie monitorowani pod kątem utraty skuteczności, co oznacza brak lub zmniejszoną odpowiedź na epoetynę alfa u pacjentów, którzy wcześniej odpowiedzieli na leczenie. Zjawisko to charakteryzuje się utrzymującym się spadkiem wartości hemoglobiny w porównaniu ze wzrostem dawki epoetyny alfa (patrz punkt 4.8).
Niektórzy pacjenci leczeni epoetyną alfa w dłuższych odstępach między dawkami (większymi niż raz w tygodniu) mogą nie utrzymywać odpowiedniego poziomu hemoglobiny (patrz punkt 5.1) i mogą wymagać zwiększenia dawki. Poziom hemoglobiny należy regularnie monitorować.
Zakrzepica dostępów naczyniowych wystąpiła u pacjentów poddawanych hemodializie, zwłaszcza u pacjentów ze skłonnością do hipotonii oraz z powikłaniami przetok tętniczo-żylnych (np. zwężenia, tętniaki itp.) U tych pacjentów zaleca się profilaktyczną kontrolę dostępu naczyniowego i profilaktykę zakrzepicy. z podaniem np. kwasu acetylosalicylowego.
W pojedynczych przypadkach obserwowano hiperkaliemię, chociaż nie ustalono związku przyczynowego. Poziom elektrolitów w surowicy należy monitorować u pacjentów z przewlekłą niewydolnością nerek. W przypadku zaobserwowania podwyższonego (lub rosnącego) stężenia potasu w surowicy, oprócz odpowiedniego leczenia hiperkaliemii, należy rozważyć przerwanie podawania epoetyny alfa do czasu wyrównania stężenia potasu w surowicy.
Często podczas hemodializy wymagane jest zwiększenie dawki heparyny ze względu na wzrost wartości hematokrytu. Jeśli dostosowanie dawki heparyny nie jest optymalne, może dojść do niedrożności dializatora. Na podstawie dotychczas dostępnych danych, wyrównanie niedokrwistości epoetyną alfa u dorosłych pacjentów z przewlekłą niewydolnością nerek niepoddawanych jeszcze dializie nie przyspiesza progresji niewydolności nerek.
Leczenie pacjentów z niedokrwistością wywołaną chemioterapią
U pacjentów z rakiem otrzymujących epoetynę alfa należy regularnie mierzyć poziom hemoglobiny aż do osiągnięcia stabilnego poziomu, a następnie wykonywać jego pomiary okresowe.
Erytropoetyny są czynnikami wzrostu, które zasadniczo stymulują produkcję czerwonych krwinek. Receptory erytropoetyny mogą ulegać ekspresji na powierzchni różnych komórek nowotworowych. Podobnie jak w przypadku wszystkich czynników wzrostu, istnieje teoretyczna obawa, że erytropoetyny mogą stymulować wzrost guza.
W niektórych kontrolowanych badaniach klinicznych nie wykazano, aby erytropoetyny poprawiały całkowity czas przeżycia lub zmniejszały ryzyko progresji nowotworu u pacjentów z niedokrwistością związaną z nowotworem.
W kontrolowanych badaniach klinicznych stosowanie epoetyny alfa i innych środków stymulujących erytropoezę (ESA) wykazało:
• zmniejszona kontrola lokoregionalna u pacjentów z zaawansowanym rakiem głowy i szyi poddawanych radioterapii w celu uzyskania stężenia hemoglobiny powyżej 14 g/dl (8,7 mmol/l);
• zmniejszone całkowite przeżycie i zwiększona śmiertelność związane z progresją choroby w ciągu 4 miesięcy u pacjentek z przerzutowym rakiem piersi otrzymujących chemioterapię po podaniu w celu uzyskania stężenia hemoglobiny w zakresie 12-14 g/dl (7,5-8,7 mmol/l);
• zwiększone ryzyko zgonu przy podawaniu w celu osiągnięcia stężenia hemoglobiny 12 g/dl (7,5 mmol/l); u pacjentów z czynną nowotworem niepoddawanych chemio- i/lub radioterapii. Leczenie środkami stymulującymi erytropoezę (ESA) nie jest wskazane w tej populacji pacjentów.
W związku z powyższym, w niektórych sytuacjach klinicznych, w leczeniu niedokrwistości u pacjentów onkologicznych preferuje się transfuzję krwi.Decyzja o podaniu rekombinowanej erytropoetyny powinna opierać się na ocenie stosunku korzyści do ryzyka z uwzględnieniem indywidualnego pacjenta. rozważenie ich szczególnego kontekstu klinicznego Czynniki, które należy wziąć pod uwagę podczas tej oceny, powinny obejmować rodzaj nowotworu i jego postęp; stopień anemii; oczekiwana długość życia, środowisko, w którym pacjent jest leczony; preferencje pacjenta (patrz punkt 5.1).
U chorych na nowotwory otrzymujących chemioterapię, 2-3-tygodniowa przerwa między podaniem a pojawieniem się czerwonych krwinek indukowanych przez ESA powinna być starannie rozważona podczas oceny stosowności leczenia epoetyną alfa (pacjenci z ryzykiem przetoczenia).
Dorośli pacjenci, którzy są kandydatami do interwencji chirurgicznych będących częścią programów autologicznego przetoczenia krwi
Należy przestrzegać wszystkich ostrzeżeń i specjalnych środków ostrożności związanych z programem autologicznego przetoczenia krwi, zwłaszcza rutynowej wymiany objętości.
Pacjenci kandydaci do dużych planowych operacji ortopedycznych
Przed operacją należy zawsze przestrzegać dobrych praktyk zarządzania krwią.
Pacjenci, którzy są kandydatami do planowego dużego zabiegu ortopedycznego, powinni otrzymać „odpowiednią profilaktykę przeciwzakrzepową, ponieważ u pacjentów poddawanych zabiegom chirurgicznym mogą wystąpić zdarzenia zakrzepowe i naczyniowe, zwłaszcza u pacjentów z towarzyszącymi chorobami sercowo-naczyniowymi. Ponadto należy zachować szczególne środki ostrożności u pacjentów z predyspozycją do rozwój zakrzepicy żył głębokich (DVT). Ponadto u pacjentów z „poziomem hemoglobiny wyjściowej > 13 g/dl” istnieje możliwość, że leczenie epoetyną alfa może wiązać się ze zwiększonym ryzykiem zdarzeń pozakrzepowych/naczyniowych. wyłączony. Dlatego epoetyny alfa nie należy stosować u pacjentów z wyjściowym stężeniem hemoglobiny > 13 g/dl.
04.5 Interakcje z innymi produktami leczniczymi i inne formy interakcji
Nie ma dowodów na to, że leczenie epoetyną alfa wpływa na metabolizm innych leków. Leki zmniejszające erytropoezę mogą osłabiać odpowiedź na epoetynę alfa.
Ponieważ cyklosporyna wiąże się z czerwonymi krwinkami, może wystąpić „interakcja z tym lekiem.
W przypadku równoczesnego podawania należy monitorować stężenie cyklosporyny we krwi, a dawkę dostosować w zależności od wzrostu hematokrytu.
Nie ma interakcji in vitro między G-CSF, GM-CSF i epoetyną alfa w zakresie różnicowania lub proliferacji hematologicznej in vitro próbek z biopsji guza.
U dorosłych pacjentek z rakiem piersi z przerzutami jednoczesne podskórne podawanie 40 000 j.m./ml epoetyny alfa z trastuzumabem w dawce 6 mg/kg nie miało wpływu na farmakokinetykę trastuzumabu.
04.6 Ciąża i laktacja
Ciąża
Nie ma odpowiednich i dobrze kontrolowanych badań u kobiet w ciąży. Badania na zwierzętach wykazały szkodliwy wpływ na reprodukcję (patrz punkt 5.3). W związku z tym epoetynę alfa należy stosować w ciąży tylko wtedy, gdy oczekiwane korzyści przewyższają potencjalne ryzyko dla płodu. Nie zaleca się stosowania epoetyny alfa u kobiet w ciąży, które kwalifikują się do zabiegu chirurgicznego biorącego udział w programie autologicznego przetoczenia krwi.
Czas karmienia
Nie wiadomo, czy egzogenna epoetyna alfa przenika do mleka ludzkiego.Epoetynę alfa należy stosować ostrożnie u kobiet karmiących piersią.Decyzję o kontynuacji/przerwaniu karmienia piersią lub kontynuacji/przerwaniu leczenia epoetyną alfa należy podjąć biorąc pod uwagę korzyści z karmienia piersią dla niemowlęcia i korzyści z leczenia epoetyną alfa dla kobiety.
Nie zaleca się stosowania epoetyny alfa u kobiet karmiących, które są kandydatami do zabiegu chirurgicznego biorących udział w programie autologicznego przetoczenia krwi.
Płodność
Nie ma badań oceniających potencjalny wpływ epoetyny alfa na płodność mężczyzn lub kobiet.
04.7 Wpływ na zdolność prowadzenia pojazdów i obsługiwania maszyn
Nie przeprowadzono badań dotyczących zdolności prowadzenia pojazdów i obsługiwania maszyn.
04.8 Działania niepożądane
Podsumowanie profilu bezpieczeństwa
Najczęstszym działaniem niepożądanym leku podczas leczenia epoetyną alfa jest zależny od dawki wzrost ciśnienia krwi lub nasilenie istniejącego wcześniej nadciśnienia tętniczego.
Zaleca się monitorowanie trendu ciśnienia krwi, szczególnie na początku leczenia (patrz punkt 4.4).
Najczęstszymi działaniami niepożądanymi leku, które wystąpiły w badaniach klinicznych z epoetyną alfa, są biegunka, nudności, wymioty, gorączka i ból głowy. Objawy grypopodobne mogą wystąpić głównie na początku leczenia.
W badaniach klinicznych dotyczących rozszerzenia zakresu dawek u dorosłych pacjentów z niewydolnością nerek niepoddawanych jeszcze dializie zgłaszano przekrwienie dróg oddechowych, w tym przypadki przekrwienia górnych dróg oddechowych, przekrwienie błony śluzowej nosa i zapalenie błony śluzowej nosa i gardła.
U pacjentów, którzy otrzymywali ESA, obserwowano zwiększoną częstość występowania zakrzepowych zdarzeń naczyniowych (TVE) (patrz punkt 4.4).
Tabela działań niepożądanych
Spośród łącznie 3262 pacjentów w 23 randomizowanych, podwójnie zaślepionych, kontrolowanych placebo lub standardowych badaniach klinicznych, ogólny profil bezpieczeństwa preparatu EPREX oceniono u 992 pacjentów z niedokrwistością. Do 4 badań z przewlekłą niewydolnością nerek włączono 228 pacjentów poddanych ICR leczonych epoetyną alfa (2 badania przed dializą [N = 131 pacjentów narażonych na ICR] i 2 badania na dializie [N = 97 pacjentów narażonych na ICR]); 1 404 pacjentów z nowotworem narażonych w 16 badaniach dotyczących niedokrwistości spowodowanej chemioterapią; 147 pacjentów narażonych w 2 badaniach dotyczących pre-oddawania krwi autologicznej i 213 pacjentów narażonych w 1 badaniu w okresie okołooperacyjnym. Działania niepożądane zgłaszane u ≥ 1% pacjentów leczonych Poniższa tabela przedstawia epoetynę alfa w tych badaniach klinicznych.
Dla różnych częstości występowania stosuje się następujące definicje: bardzo często (≥ 1/10), często (≥ 1/100 do
Opis wybranych działań niepożądanych
Zgłaszano reakcje nadwrażliwości, w tym przypadki wysypki (w tym pokrzywki), reakcje anafilaktyczne i obrzęk naczynioruchowy.
Kryzysy nadciśnieniowe z encefalopatią i drgawkami, które wymagały natychmiastowej pomocy lekarskiej i intensywnej opieki medycznej, występowały podczas leczenia epoetyną alfa nawet u pacjentów z poprzednio prawidłowym lub niskim ciśnieniem krwi. Szczególną uwagę należy zwrócić na kłucia przypominające migrenę jako możliwy znak ostrzegawczy (patrz punkt 4.4).
Bardzo rzadko zgłaszano aplazję wybiórczą krwinek czerwonych, w której pośredniczą przeciwciała u:
Populacja pediatryczna z przewlekłą niewydolnością nerek podczas hemodializy
Ekspozycja pacjentów pediatrycznych z przewlekłą niewydolnością nerek poddawanych hemodializie w badaniach klinicznych i po wprowadzeniu produktu do obrotu jest ograniczona. W populacji tej nie zgłoszono żadnych specyficznych działań niepożądanych z populacji pediatrycznej niewymienionych w powyższej tabeli ani żadnych działań niepożądanych typowych dla choroby podstawowej.
Zgłaszanie podejrzewanych działań niepożądanych
Zgłaszanie podejrzewanych działań niepożądanych występujących po dopuszczeniu produktu leczniczego do obrotu jest ważne, ponieważ umożliwia ciągłe monitorowanie stosunku korzyści do ryzyka produktu leczniczego. Osoby należące do fachowego personelu medycznego są proszone o zgłaszanie wszelkich podejrzewanych działań niepożądanych za pośrednictwem krajowego systemu zgłaszania. „adres http: //www.agenziafarmaco.gov.it/it/responsabili.
04.9 Przedawkowanie
Margines terapeutyczny epoetyny alfa jest bardzo szeroki. Przedawkowanie epoetyny alfa może wywołać skutki, które są przedłużeniem działania farmakologicznego hormonu.W przypadku stwierdzenia nadmiernie wysokiego stężenia hemoglobiny można wykonać upuszczanie krwi.
W zależności od stanu pacjenta należy zapewnić dodatkową opiekę podtrzymującą.
05.0 WŁAŚCIWOŚCI FARMAKOLOGICZNE
05.1 Właściwości farmakodynamiczne
Grupa farmakoterapeutyczna: leki przeciw niedokrwistości, kod ATC: B03XA01.
Mechanizm akcji
Erytropoetyna (EPO) jest hormonem glikoproteinowym wytwarzanym głównie przez nerki w odpowiedzi na niedotlenienie i jest kluczowym regulatorem produkcji czerwonych krwinek (RBC). „EPO bierze udział we wszystkich fazach rozwoju erytroidów i ma swój główny wpływ na poziom prekursorów erytroidów. Po związaniu EPO ze swoim receptorem na powierzchni komórki aktywuje szlaki transdukcji sygnału, które zakłócają apoptozę i stymulują proliferację komórek erytroidalnych. ludzka erytropoetyna (epoetyna alfa), wyrażona w komórkach jajnika chomika chińskiego, ma sekwencję 165 aminokwasów identyczną z sekwencją ludzkiej EPO w moczu; te 2 są nie do odróżnienia na podstawie testów funkcjonalnych Pozorna masa cząsteczkowa erytropoetyny wynosi od 32 000 do 40 000 daltony.
Erytropoetyna jest czynnikiem wzrostu, który przede wszystkim stymuluje produkcję erytrocytów.Receptory erytropoetyny mogą ulegać ekspresji na powierzchni różnych komórek nowotworowych.
Efekty farmakodynamiczne
Zdrowi wolontariusze
Po podaniu pojedynczych dawek epoetyny alfa (20 000 do 160 000 jm podskórnie) zaobserwowano odpowiedź zależną od dawki dla badanych markerów farmakodynamicznych, w tym: retikulocytów, krwinek czerwonych i hemoglobiny. Zaobserwowano zdefiniowany profil stężenie-czas ze szczytem i powrotem do wartości wyjściowej dla zmian odsetka retikulocytów. Mniej zdefiniowany profil zaobserwowano dla erytrocytów i hemoglobiny. Ogólnie rzecz biorąc, wszystkie markery farmakodynamiczne wzrastały liniowo wraz z dawkami osiągającymi maksymalną odpowiedź przy maksymalnych poziomach dawki.
W dodatkowych badaniach farmakodynamicznych oceniano 40 000 jm raz w tygodniu w porównaniu do 150 jm/kg 3 razy w tygodniu. Pomimo różnic w profilach stężenie-czas, odpowiedź farmakodynamiczna (mierzona procentowymi zmianami w retikulocytach, hemoglobinie i całkowitej liczbie czerwonych krwinek) była podobna dla tych schematów. W dodatkowych badaniach porównano schemat 40 000 jm epoetyny alfa raz w tygodniu z dawkami podawanymi co dwa tygodnie w zakresie od 80 000 do 120 000 jm podskórnie. Ogólnie rzecz biorąc, na podstawie wyników tych badań farmakodynamicznych u zdrowych osób, schemat dawkowania 40 000 j.m. raz w tygodniu wydaje się być skuteczniejszy w wytwarzaniu krwinek czerwonych niż schematy dwutygodniowe, pomimo podobieństwa w wytwarzaniu retikulocytów. reżimy dwutygodniowe.
Przewlekłą niewydolność nerek
Wykazano, że epoetyna alfa stymuluje erytropoezę u pacjentów z niedokrwistością i przewlekłą niewydolnością nerek, w tym u pacjentów dializowanych i przed dializą. Pierwszym dowodem odpowiedzi na epoetynę alfa jest zwiększenie liczby retikulocytów w ciągu 10 dni, a następnie zwiększenie liczby krwinek czerwonych, hemoglobiny i hematokrytu, zwykle w ciągu 2-6 tygodni. Odpowiedź hemoglobiny różni się w zależności od pacjenta i może na nią wpływać złogi żelaza oraz współistniejące problemy zdrowotne.
Niedokrwistość wywołana chemioterapią
Wykazano, że epoetyna alfa podawana 3 razy w tygodniu lub raz w tygodniu zwiększa stężenie hemoglobiny i zmniejsza potrzebę transfuzji po pierwszym miesiącu leczenia u pacjentów z niedokrwistością nowotworową otrzymujących chemioterapię.
W badaniu porównującym schematy dawkowania 150 j.m./kg 3 razy w tygodniu i 40 000 j.m. raz w tygodniu u osób zdrowych i pacjentów z nowotworem i niedokrwistością, profile czasowe zmian procentowych w retikulocytach, hemoglobinie i całkowitej liczbie czerwonych krwinek były podobne między dwa schematy dawkowania u osób zdrowych i chorych na raka z niedokrwistością. AUC odpowiednich parametrów farmakodynamicznych były podobne między schematami dawkowania 150 jm/kg 3 razy w tygodniu i 40 000 jm raz w tygodniu zarówno u zdrowych, jak i niedokrwistości pacjentów z rakiem.
Dorośli pacjenci chirurgiczni w programie autologicznej predonacji
Wykazano, że epoetyna alfa stymuluje wytwarzanie krwinek czerwonych w celu zwiększenia autologicznego pobierania krwi i ograniczenia spadku stężenia hemoglobiny u dorosłych pacjentów zakwalifikowanych do dużych planowych operacji chirurgicznych, u których nie przewiduje się pełnego przechowywania przedoperacyjnego w okresie okołooperacyjnym. Największe efekty zaobserwowano u pacjentów z niskim poziomem hemoglobiny (≤ 13 g/dl).
Leczenie dorosłych pacjentów zakwalifikowanych do poważnej planowej operacji ortopedycznej
Wykazano, że u pacjentów zakwalifikowanych do dużej planowej operacji ortopedycznej ze wstępnym leczeniem hemoglobiny > 10 do ≤ 13 g / dl epoetyna alfa zmniejsza ryzyko otrzymywania przetoczeń allogenicznych i przyspiesza regenerację erytroidów (podwyższony poziom hemoglobiny, hematokrytu i liczby retikulocytów).
Skuteczność kliniczna i bezpieczeństwo
Przewlekłą niewydolność nerek
Epoetynę alfa oceniano w badaniach klinicznych u dorosłych pacjentów z niedokrwistością z CRF, w tym pacjentów poddawanych hemodializie i przed dializą, w celu leczenia niedokrwistości i utrzymania hematokrytu w zakresie stężeń od 30 do 36%.
W badaniach klinicznych przy dawkach początkowych 50-150 j.m./kg trzy razy w tygodniu około 95% wszystkich pacjentów odpowiedziało klinicznie istotnym wzrostem hematokrytu.Po około dwóch miesiącach leczenia prawie wszyscy pacjenci otrzymali transfuzję. osiągnięto docelową wartość hematokrytu, dawkę podtrzymującą dostosowywano dla każdego pacjenta.
W trzech głównych badaniach klinicznych u dorosłych pacjentów dializowanych średnia dawka podtrzymująca potrzebna do utrzymania hematokrytu między 30 a 36% wynosiła około 75 jm/kg 3 razy w tygodniu.
W podwójnie zaślepionym, wieloośrodkowym, kontrolowanym placebo badaniu jakości życia pacjentów z przewlekłą niewydolnością nerek poddawanych hemodializie wykazano klinicznie i statystycznie istotną poprawę u pacjentów leczonych epoetyną alfa w porównaniu z grupą placebo, mierząc zmęczenie, objawy fizyczne, związki oraz depresja (Kwestionariusz Chorób Nerek) po sześciu miesiącach terapii. Pacjenci w grupie otrzymującej epoetynę alfa zostali włączeni do otwartego przedłużonego badania, które wykazało, że poprawa ich jakości życia utrzymywała się przez dodatkowe 12 miesięcy.
Dorośli pacjenci z niewydolnością nerek niepoddani jeszcze dializie
W badaniach klinicznych u pacjentów z przewlekłą niewydolnością nerek niepoddawanych dializie, leczonych epoetyną alfa, średni czas trwania leczenia wynosił prawie pięć miesięcy. Pacjenci ci zareagowali na leczenie epoetyną alfa w sposób podobny do obserwowanego u pacjentów dializowanych. Pacjenci z CRF nie poddawani dializie wykazywali zależność od dawki i stały wzrost hematokrytu, gdy epoetyna alfa była podawana zarówno dożylnie, jak i podskórnie. Podobne tempo wzrostu hematokrytu zaobserwowano, gdy podawano epoetynę alfa w obu przypadkach. Ponadto, dawki epoetyny alfa od 75 do 150 jm / kg Wykazano, że na tydzień utrzymuje hematokryt na poziomie 36 do 38% przez okres do sześciu miesięcy.
W 2 badaniach z wydłużonymi przerwami między dawkami leku EPREX (3 razy w tygodniu, raz w tygodniu, raz na dwa tygodnie i raz na cztery tygodnie) niektórzy pacjenci z dłuższymi przerwami między dawkami nie utrzymywali odpowiednich poziomów hemoglobiny i spełniali ustalone kryteria odstawienia (0% raz w tygodniu, 3,7% raz na 2 tygodnie i 3,3% w grupach raz na 4 tygodnie).
W prospektywnym randomizowanym badaniu (CHOIR) oceniono 1432 pacjentów z niedokrwistością z przewlekłą niewydolnością nerek niepoddawanych dializie. Pacjentom przypisano leczenie epoetyną alfa w celu utrzymania poziomu hemoglobiny 13,5 g/dl (powyżej zalecanego poziomu hemoglobiny) lub 11,3 g/dl.Poważne zdarzenie sercowo-naczyniowe (zgon, zawał mięśnia sercowego, udar lub hospitalizacja z powodu zastoinowej niewydolności serca) , wystąpiło wśród 125 (18%) z 715 pacjentów w grupie z wyższą hemoglobiną w porównaniu do 97 (14%) wśród 717 pacjentów w grupie z niższą hemoglobiną (procent ryzyka [HR] 1,3, 95% CI: 1,0, 1,7, p = 0,03).
Zbiorcze retrospektywne analizy badań klinicznych ESA przeprowadzono u pacjentów z przewlekłą niewydolnością nerek (u pacjentów dializowanych i niedializowanych, pacjentów z cukrzycą i bez cukrzycy). Obserwowano trend w kierunku zwiększonego szacowanego ryzyka zgonów z jakiejkolwiek przyczyny, zdarzeń sercowo-naczyniowych i naczyniowo-mózgowych związanych z większymi skumulowanymi dawkami ESA, niezależnie od stanu cukrzycy lub dializy pacjenta (patrz punkt 4.2 i punkt 4.4).
Leczenie pacjentów z niedokrwistością wywołaną chemioterapią
Epoetynę alfa oceniano w badaniach klinicznych u dorosłych pacjentów z niedokrwistością nowotworową z guzami limfoidalnymi i litymi oraz u pacjentów otrzymujących różne schematy chemioterapii, w tym schematy zawierające platynę i nie zawierające platyny. W badaniach tych wykazano, że epoetyna alfa podawana 3 razy w tygodniu i raz w tygodniu zwiększa stężenie hemoglobiny i zmniejsza potrzebę transfuzji po pierwszym miesiącu leczenia u pacjentów z niedokrwistością nowotworową. W niektórych badaniach po fazie podwójnie ślepej próby następowała faza otwartej próby, podczas której wszyscy pacjenci otrzymywali epoetynę alfa i obserwowano utrzymanie efektu.
Dostępne dane sugerują, że pacjenci z nowotworami złośliwymi układu krwiotwórczego i guzami litymi odpowiadają równoważnie na terapię epoetyną alfa, a pacjenci z naciekiem nowotworu szpiku kostnego lub bez nacieku kostnego odpowiadają równoważnie na terapię epoetyną alfa. Porównywalną intensywność chemioterapii w grupach epoetyny alfa i placebo w badaniach dotyczących chemioterapii wykazywał podobny obszar pod krzywą czasu neutrofili u pacjentów leczonych epoetyną alfa i placebo, a także podobny odsetek pacjentów w leczonych grupach. z grupami otrzymującymi epoetynę alfa i placebo, których bezwzględna liczba neutrofili spadła poniżej 1000 i 500 komórek/mcL
W prospektywnym, podwójnie zaślepionym, randomizowanym, kontrolowanym placebo badaniu obejmującym 375 pacjentów z niedokrwistością i guzami nieszpikowymi, otrzymujących chemioterapię nieopartą na platynie, znaczne zmniejszenie objawów związanych z niedokrwistością (takich jak astenia, zmęczenie i obniżone ciśnienie krwi) aktywność), mierzoną następującymi skalami oceny: skala ogólna funkcjonalnej oceny terapii nowotworowej-niedokrwistości (FACT-An); FACT – Skala zmęczenia i Cancer Linear Analogue Scale (CLAS).
Dwa inne badania z randomizacją i placebo przeprowadzone na mniejszej liczbie pacjentów nie wykazały żadnej poprawy parametrów jakości życia według skal EORTC-QLQ-C30 i CLAS.
Przeżycie i progresję guza zbadano w pięciu dużych badaniach kontrolowanych z udziałem łącznie 2833 pacjentów, w tym czterech podwójnie ślepych, kontrolowanych placebo i jednego otwartego. Do badań włączono zarówno pacjentów leczonych chemioterapią (dwa badania), jak i pacjentów, u których stosowanie ŚSE nie było wskazane: pacjenci z nowotworem niedokrwistości nie otrzymujący chemioterapii oraz pacjenci z rakiem głowy i szyi otrzymujący radioterapię Pożądane stężenie radioterapii hemoglobina u dwóch pacjentów w badaniach wynosił > 13 g / dl, w pozostałych trzech badaniach wynosił od 12 do 14 g / dl W badaniu otwartym nie stwierdzono różnicy w przeżywalności między pacjentami leczonymi rekombinowaną ludzką erytropoetyną a grupą kontrolną. W czterech kontrolowanych badaniach"współczynnik ryzyka dla przeżycia całkowitego wahał się od 1,25 do 2,47 na korzyść grupy kontrolnej. Badania te wykazały niewyjaśniony statystycznie istotny wzrost śmiertelności u pacjentów z niedokrwistością nowotworową leczonych rekombinowaną ludzką erytropoetyną w porównaniu z grupą kontrolną. Całkowity wynik przeżycia pacjentów leczonych rekombinowaną ludzką erytropoetyną w porównaniu z grupą kontrolną nie mógł być zadowalająco wyjaśniony różnicą w częstości występowania zakrzepicy i związanych z nią powikłań.
Przeprowadzono również analizę na poziomie pacjenta ponad 13 900 pacjentów z nowotworami (chemio-, radio-, radiochemio- lub nieleczonych), którzy uczestniczyli w 53 kontrolowanych badaniach klinicznych dotyczących różnych epoetyn. Metaanaliza danych dotyczących przeżycia całkowitego wykazała punktowy współczynnik ryzyka wynoszący 1,06 na korzyść grupy kontrolnej (95% CI: 1,00; 1,12; 53 badania i 13933 pacjentów), podczas gdy w przypadku pacjentów z nowotworami złośliwymi otrzymujących chemioterapię całkowite przeżycie w zakresie współczynnik ryzyka wynosił 1,04 (95% CI: 0,97, 1,11; 38 badań i 10441 pacjentów). Ponadto metaanalizy konsekwentnie wskazują na znaczne zwiększenie względnego ryzyka zdarzeń zakrzepowo-zatorowych u pacjentów z nowotworem otrzymujących rekombinowane ludzkie erytropoetyny (patrz punkt 4.4).
Program predonacji autologicznej
Wpływ epoetyny alfa na ułatwienie autologicznego oddawania krwi u pacjentów z niskim hematokrytem (≤ 39% i bez wyraźnej niedokrwistości spowodowanej niedoborem żelaza) zakwalifikowanych do poważnej operacji ortopedycznej oceniano w podwójnym badaniu kontrolowanym placebo. -ślepe badanie kontrolowane placebo z udziałem 55 pacjentów.
W badaniu z podwójnie ślepą próbą pacjenci otrzymywali epoetynę alfa 600 j.m./kg lub placebo dożylnie raz na dobę co 3 lub 4 dni przez 3 tygodnie (w sumie 6 dawek). Przeciętnie pacjenci leczeni epoetyną alfa byli w stanie wstępnie zdeponować znacznie więcej jednostek krwi (4,5 jednostki) niż pacjenci otrzymujący placebo (3,0 jednostki).
W badaniu z pojedynczą ślepą próbą pacjenci otrzymywali epoetynę alfa 300 j.m./kg lub 600 j.m./kg lub placebo dożylnie raz na dobę co 3 lub 4 dni przez 3 tygodnie (łącznie 6 dawek). Pacjenci leczeni epoetyną alfa byli również w stanie wstępnie odłożyć znaczne dodatkowe jednostki krwi (epoetyna alfa 300 j.m./kg = 4,4 jednostki; epoetyna alfa 600 j.m./kg = 4,7 jednostki) w porównaniu z pacjentami otrzymującymi placebo (2,9 jednostki).
Terapia epoetyną alfa zmniejszyła ryzyko ekspozycji allogenicznej we krwi o 50% w porównaniu z pacjentami nieleczonymi epoetyną alfa.
Duża planowa operacja ortopedyczna
Wpływ epoetyny alfa (300 j.m./kg lub 100 j.m./kg) na ekspozycję na allogeniczne transfuzje krwi oceniano w podwójnie zaślepionym, kontrolowanym placebo badaniu klinicznym z udziałem dorosłych pacjentów z niedoborem żelaza oczekujących na operację. lub operacja kolana Epoetynę alfa podawano podskórnie przez 10 dni przed operacją, w dniu operacji i przez cztery dni po operacji. Pacjenci byli klasyfikowani według ich wyjściowej hemoglobiny (≤ 10 g / dl, > 10 do ≤ 13 g / dl i > 13 g / dl).
Epoetyna alfa 300 j.m./kg istotnie zmniejszała ryzyko przetoczeń allogenicznych u pacjentów z hemoglobiną przed leczeniem z > 10 do ≤ 13 g/dl. 16% pacjentów leczonych epoetyną alfa 300 j.m./kg, 23% epoetyną alfa 100 j.m./kg i 45% placebo wymagało transfuzji.
Otwarte badanie w grupach równoległych z udziałem dorosłych pacjentów z niedoborem żelaza i hemoglobiną przed leczeniem ≥ 10 do ≤ 13 g / dl, którzy czekali na ortopedyczną operację stawu biodrowego lub kolanowego, porównywało leczenie epoetyną alfa 300 j.m. / kg codziennie przez 10 dni przed zabiegiem, w w dniu zabiegu i przez cztery dni po zabiegu, z epoetyną alfa 600 j.m./kg podskórnie raz w tygodniu przez 3 tygodnie przed zabiegiem oraz w dniu zabiegu.
Od leczenia wstępnego do przedoperacyjnego średni wzrost stężenia hemoglobiny w grupie tygodniowej 600 jm/kg (1,44 g/dl) był dwukrotnie wyższy niż obserwowany w grupie 300 jm/kg dziennie (0,73 g/dl). Średnie poziomy hemoglobiny były podobne w obu leczonych grupach przez cały okres pooperacyjny.
Odpowiedź erytropoetyczna obserwowana w obu leczonych grupach prowadziła do podobnych szybkości transfuzji (16% w grupie 600 IU/kg/tydzień i 20% w grupie 300 IU/kg/dzień)
Populacja pediatryczna
Przewlekłą niewydolność nerek
Epoetynę alfa oceniano w otwartym, nierandomizowanym, 52-tygodniowym badaniu klinicznym z otwartym zakresem dawek u dzieci i młodzieży z przewlekłą niewydolnością nerek poddawanych hemodializie. Średni wiek pacjentów włączonych do badania wynosił 11,6 lat (zakres od 0,5 do 20,1 lat).
Epoetynę alfa podawano dożylnie w dawce 75 j.m./kg/tydzień w 2 lub 3 dawkach podzielonych po dializie, miareczkowanych od 75 j.m./kg/tydzień w odstępach 4-tygodniowych (maksymalnie 300 j.m./kg/tydzień). aby osiągnąć wzrost stężenia hemoglobiny o 1 g/dl/miesiąc Pożądany poziom hemoglobiny wynosił od 9,6 do 11,2 g/dl Osiemdziesiąt jeden procent pacjentów osiągnęło docelowy poziom hemoglobiny. Mediana czasu do miejsca przeznaczenia wyniosła 11 tygodni, mediana dawki do miejsca przeznaczenia wyniosła 150 IU/kg/tydzień. Spośród pacjentów, którzy osiągnęli cel, 90% zrobiło to przy schemacie dawkowania 3 razy w tygodniu.
Po 52 tygodniach 57% pacjentów pozostało w badaniu otrzymując średnią dawkę 200 jm/kg/tydzień.
05.2 Właściwości farmakokinetyczne
Wchłanianie
Po podaniu podskórnym maksymalne stężenie epoetyny alfa w surowicy występuje pomiędzy 12 a 18 godziną po podaniu. Nie stwierdzono kumulacji po podaniu wielokrotnych tygodniowych dawek 600 jm/kg podawanych podskórnie.
Całkowita biodostępność epoetyny alfa podawanej podskórnie u zdrowych osób wynosi około 20%.
Dystrybucja
Średnia objętość dystrybucji wynosi 49,3 ml/kg po dożylnym podaniu zdrowym osobom 50 i 100 jm/kg. Po dożylnym podaniu epoetyny alfa osobom z przewlekłą niewydolnością nerek objętość dystrybucji wahała się od 57-107 ml/kg po podaniu jednorazowym (12 j.m./kg) do 42-64 ml/kg po podaniu wielokrotnym (48-192 j.m./kg). / kg). W konsekwencji objętość dystrybucji jest nieco większa niż objętość osocza.
Eliminacja
Okres półtrwania epoetyny alfa po dożylnym podaniu wielokrotnych dawek u zdrowych osób wynosi około 4 godziny, a po podaniu podskórnym szacuje się na około 24 godziny u zdrowych osób.
Średnia wartość CL/F dla schematów 150 jm/kg 3 razy w tygodniu i 40 000 jm raz w tygodniu u zdrowych osób wynosiła odpowiednio 31,2 i 12,6 ml/h/kg. Średnia wartość CL/F dla schematów 150 jm/kg 3 razy w tygodniu i 40 000 jm raz w tygodniu u pacjentów z niedokrwistością raka wynosiła odpowiednio 45,8 i 11,3 ml/h/kg. U większości osób z niedokrwistością, otrzymujących chemioterapię cykliczną, CL/F był niższy po podskórnych dawkach 40 000 IU raz w tygodniu i 150 IU/kg 3 razy w tygodniu w porównaniu z wartościami dla osób zdrowych.
Liniowość / nieliniowość
U zdrowych osób obserwowano proporcjonalny do dawki wzrost stężenia epoetyny alfa w surowicy po podaniu dożylnym 150 i 300 jm/kg 3 razy w tygodniu. Pojedyncze podskórne dawki 300-2400 jm/kg epoetyny alfa powodowały liniową zależność między średnim Cmax a dawką oraz między średnią AUC a dawką. Zaobserwowano odwrotną zależność między luzpozorna i dawka u zdrowych osób.
W badaniach dotyczących wydłużenia odstępu między dawkami (40 000 j.m. raz w tygodniu oraz 80 000, 100 000 i 120 000 j.m. co dwa tygodnie) zaobserwowano liniową, ale nieproporcjonalną do dawki zależność między średnią wartością Cmax i dawką el ” Średnia AUC i dawką w stanie stacjonarnym .
Zależność farmakokinetyczna/farmakodynamiczna
Epoetyna alfa wykazuje zależny od dawki wpływ na parametry hematologiczne, który jest niezależny od drogi podania.
Populacja pediatryczna
Po wielokrotnym dożylnym podaniu epoetyny alfa u dzieci z przewlekłą niewydolnością nerek zgłaszano okres półtrwania wynoszący około 6,2-8,7 h. Profil farmakokinetyczny epoetyny alfa u dzieci i młodzieży wydaje się być podobny do profilu u dorosłych.
Niewydolność nerek
U pacjentów z przewlekłą niewydolnością nerek okres półtrwania epoetyny alfa podawanej dożylnie jest nieco dłuższy, około 5 godzin, w porównaniu z osobami zdrowymi.
05.3 Przedkliniczne dane o bezpieczeństwie
W badaniach toksykologicznych po podaniu wielokrotnym u psów i szczurów, ale nie u małp, terapia epoetyną alfa była związana z subklinicznym stanem zwłóknienia szpiku kostnego. Zwłóknienie szpiku kostnego jest znanym powikłaniem przewlekłej niewydolności nerek u ludzi i może być związane z wtórną nadczynnością przytarczyc lub być spowodowane nieznanymi czynnikami. W badaniu z udziałem pacjentów hemodializowanych leczonych epoetyną alfa przez 3 lata częstość występowania zwłóknienia szpiku kostnego nie była większa niż w grupie kontrolnej pacjentów dializowanych nieleczonych epoetyną alfa.
Epoetyna alfa nie wywołuje mutacji genów u bakterii (test Amesa), aberracji chromosomowych w komórkach ssaków, mikrojąder u myszy ani mutacji genów w locus HGPRT.
Nie przeprowadzono długoterminowych badań rakotwórczości. Sprzeczne wyniki w literaturze na podstawie danych in vitro z próbek ludzkich guzów sugerują, że erytropoetyny mogą odgrywać rolę w proliferacji guza, co ma niepewne znaczenie w praktyce klinicznej.
W hodowlach komórkowych ludzkich komórek szpiku kostnego epoetyna alfa specyficznie stymuluje erytropoezę i nie obejmuje leukopoezy.Nie zaobserwowano cytotoksycznego działania epoetyny alfa na komórki szpiku kostnego.
W badaniach na zwierzętach wykazano, że epoetyna alfa podawana w cotygodniowych dawkach około 20-krotności zalecanej dawki tygodniowej u ludzi zmniejsza masę ciała płodu, opóźnia kostnienie i zwiększa śmiertelność płodów. Zmiany te uważa się za wtórne do zmniejszenia masy ciała matki, a ich znaczenie dla ludzi w dawkach terapeutycznych nie jest znane.
06.0 INFORMACJE FARMACEUTYCZNE
06.1 Zaróbki
Polisorbat 80
Glicyna
Woda do wstrzykiwań.
Dihydrat jednozasadowego fosforanu sodu
Dihydrat dwuzasadowego fosforanu sodu
Chlorek sodu
06.2 Niezgodność
Ze względu na brak badań zgodności produktu leczniczego nie wolno mieszać z innymi produktami.
06.3 Okres ważności
18 miesięcy.
06.4 Specjalne środki ostrożności przy przechowywaniu
Przechowywać w lodówce (2°C - 8°C). Ten zakres temperatur musi być zapewniony do czasu podania pacjentowi.
Przechowywać w oryginalnym opakowaniu, aby chronić produkt przed światłem.
Nie zamrażać ani nie wstrząsać.
W przypadku stosowania ambulatoryjnego lek można wyjąć z lodówki bez wymiany przez maksymalnie 3 dni w temperaturze nieprzekraczającej 25°C. Jeśli lek nie został zużyty do końca tego okresu, należy go zutylizować.
06.5 Rodzaj opakowania bezpośredniego i zawartość opakowania
0,3 ml (3000 j.m.) roztworu do wstrzykiwań w ampułko-strzykawce (szkło typu I) z tłokiem (guma pokryta teflonem) i igłą z obudową (guma z powłoką polipropylenową) i zabezpieczeniem igły PROTECS (poliwęglan) przymocowanym do strzykawki - opakowanie 1 szt.
0,4 ml (4 000 j.m.) w ampułko-strzykawce (szkło typu I) z tłokiem (guma pokryta teflonem) i igłą z obudową (gumową z powłoką polipropylenową) i urządzeniem zabezpieczającym igłę PROTECS (poliwęglan) dołączonym do strzykawki – opakowanie zawiera 1.
0,5 ml (5000 j.m.) roztworu do wstrzykiwań w ampułko-strzykawce (szkło typu I) z tłokiem (guma pokryta teflonem) i igłą z obudową (guma z powłoką polipropylenową) i zabezpieczeniem igły PROTECS (poliwęglan) przymocowanym do strzykawki - opakowanie 1 szt.
0,6 ml (6000 j.m.) roztworu do wstrzykiwań w ampułko-strzykawce (szkło typu I) z tłokiem (guma pokryta teflonem) i igłą z obudową (guma pokryta polipropylenem) i zabezpieczeniem igły PROTECS (poliwęglan) przymocowanym do strzykawki - opakowanie 1 szt.
0,8 ml (7 000 j.m.) roztworu do wstrzykiwań w ampułko-strzykawce (szkło typu I) z tłokiem (guma pokryta teflonem) i igłą z obudową (guma z powłoką polipropylenową) i zabezpieczeniem igły PROTECS (poliwęglan) przymocowanym do strzykawki - opakowanie 1 szt.
1,0 ml (10 000 j.m.) roztworu do wstrzykiwań w ampułko-strzykawce (szkło typu I) z tłokiem (guma pokryta teflonem) i igłą z obudową (guma z powłoką polipropylenową) i zabezpieczeniem igły PROTECS (poliwęglan) przymocowanym do strzykawki - opakowanie 1 szt.
06.6 Instrukcje użytkowania i obsługi
Produktu nie wolno używać i należy go wyrzucić:
• Jeśli plomba jest zerwana;
• Jeśli roztwór jest zabarwiony lub zawiera cząstki;
• Jeżeli wystąpiło lub podejrzewa się zamarznięcie;
• W przypadku awarii lodówki.
Produkt jednorazowy. Nie podawać więcej niż jednej dawki na strzykawkę po pobraniu niechcianej ilości roztworu ze strzykawki. Patrz punkt 3. Jak stosować EPREX (instrukcja wstrzykiwania) w ulotce dołączonej do opakowania.
Ampułko-strzykawki są wyposażone w zabezpieczenie igły PROTECS, aby zapobiec ryzyku ukłucia się igłą.
Niewykorzystany lek i odpady pochodzące z tego leku należy usunąć zgodnie z lokalnymi przepisami.
07.0 PODMIOT POZWOLENIA NA DOPUSZCZENIE DO OBROTU
Janssen-Cilag SpA, via M.Buonarroti, 23 - 20093 Cologno Monzese (MI)
08.0 NUMER POZWOLENIA NA DOPUSZCZENIE DO OBROTU
027015167 - „10 000 j.m./ml roztwór do wstrzykiwań w ampułko-strzykawce” 1 strzykawka 3000 j.m./0,3 ml
027015179 - „10 000 j.m./ml roztwór do wstrzykiwań w ampułko-strzykawce” 1 strzykawka 4000 j.m./0,4 ml
027015231 - „10 000 j.m./ml roztwór do wstrzykiwań w ampułko-strzykawce” 1 strzykawka 5000 j.m./0,5 ml
027015243 – „10 000 j.m./ml roztwór do wstrzykiwań w ampułko-strzykawce” 1 strzykawka 6 000 j.m./0,6 ml
027015268 - „10 000 j.m./ml roztwór do wstrzykiwań w ampułko-strzykawce” 1 strzykawka 8000 j.m./0,8 ml
027015181 - „10 000 j.m./ml roztwór do wstrzykiwań w ampułko-strzykawce” 1 strzykawka 10 000 j.m./1 ml
09.0 DATA PIERWSZEGO ZEZWOLENIA LUB PRZEDŁUŻENIA ZEZWOLENIA
maj 1989
Odnowienie AIC: 4 sierpnia 2008 r.
10.0 DATA ZMIAN TEKSTU
08/2015