Składniki aktywne: Dabigatran (eteksylan dabigatranu)
Pradaxa 150 mg kapsułki twarde
Ulotki informacyjne Pradaxa są dostępne dla wielkości opakowań:- Pradaxa 75 mg kapsułki twarde
- Pradaxa 110 mg kapsułki twarde
- Pradaxa 150 mg kapsułki twarde
Dlaczego stosuje się Pradaxę? Po co to jest?
Pradaxa to lek zawierający substancję czynną eteksylan dabigatranu. Blokuje działanie substancji w organizmie, która bierze udział w tworzeniu skrzepów krwi.
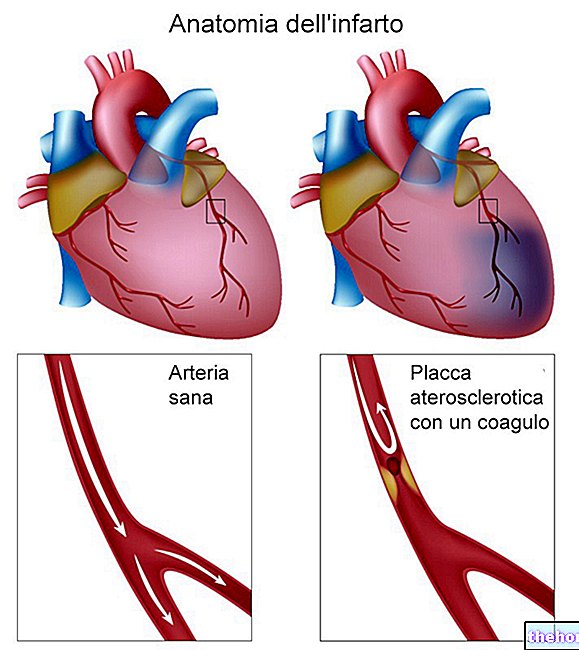
Pradaxa jest lekiem stosowanym w celu zmniejszenia ryzyka naczyń krwionośnych w mózgu lub reszcie ciała zablokowanych przez tworzenie się skrzepów krwi u dorosłych pacjentów ze zmienionym rytmem serca (migotanie przedsionków) oraz z dodatkowymi czynnikami ryzyka. Pradaxa to lek rozrzedzający krew, zmniejszający ryzyko powstawania zakrzepów krwi.
Pradaxa jest lekiem stosowanym w leczeniu zakrzepów krwi w żyłach nóg i płuc oraz zapobieganiu nawrotom zakrzepów krwi w żyłach nóg i płuc.
Przeciwwskazania Kiedy nie należy stosować leku Pradaxa
Nie należy przyjmować leku Pradaxa
- jeśli pacjent ma uczulenie na eteksylan dabigatranu lub którykolwiek z pozostałych składników tego leku (wymienionych w punkcie 6).
- jeśli czynność nerek jest poważnie osłabiona;
- jeśli masz ciągłe krwawienie.
- jeśli masz uraz narządu, który zwiększa ryzyko ciężkiego krwawienia.
- jeśli u pacjenta występuje zwiększona skłonność do krwawień, która może być wrodzona, o nieznanej przyczynie lub z powodu innych leków.
- jeśli u pacjenta występuje poważne upośledzenie czynności wątroby lub choroba wątroby, która może w jakiś sposób spowodować śmierć.
- jeśli pacjent przyjmuje ketokonazol lub itrakonazol doustnie, leki stosowane w leczeniu zakażeń grzybiczych.
- jeśli pacjent przyjmuje cyklosporynę, lek zapobiegający epizodom odrzucenia po przeszczepieniu narządu.
- jeśli pacjent przyjmuje dronedaron, lek zapobiegający nawrotom zaburzeń rytmu serca.
- jeśli pacjent przyjmuje leki zapobiegające tworzeniu się zakrzepów krwi (np. warfaryna, rywaroksaban, apiksaban lub heparyna), z wyjątkiem sytuacji, gdy zmienia się jeden lek przeciwzakrzepowy na inny lub gdy założony jest cewnik do żyły tętniczej i przyjmuje przez niego heparynę w celu utrzymania go otwartego.
- jeśli pacjentowi wszczepiono sztuczną zastawkę serca.
Środki ostrożności dotyczące stosowania Informacje ważne przed zastosowaniem leku Pradaxa
Przed przyjęciem leku Pradaxa należy porozmawiać z lekarzem. Konieczne może być również skontaktowanie się z lekarzem podczas leczenia lekiem Pradaxa w przypadku wystąpienia objawów lub konieczności przeprowadzenia zabiegu chirurgicznego. Należy poinformować lekarza, jeśli masz lub cierpiałeś na jakiekolwiek schorzenie lub chorobę, szczególnie te wymienione na poniższej liście:
- jeśli u pacjenta występuje choroba wątroby związana z nieprawidłowymi wynikami badań krwi, nie zaleca się stosowania leku Pradaxa.
- jeśli u pacjenta występuje zwiększone ryzyko krwawienia, które może wystąpić w następujących sytuacjach:
- jeśli ostatnio wystąpiło krwawienie.
- jeśli pacjent miał biopsję (chirurgiczne usunięcie tkanki) w poprzednim miesiącu.
- jeśli pacjent doznał poważnych obrażeń (np. złamania kości, urazu głowy lub jakiegokolwiek urazu wymagającego zabiegu chirurgicznego).
- jeśli u pacjenta występuje zapalenie przełyku lub żołądka.
- jeśli masz problemy z cofaniem się soku żołądkowego do przełyku.
- jeśli pacjent przyjmował leki, które mogą zwiększać ryzyko krwawienia, takie jak aspiryna (kwas acetylosalicylowy), klopidogrel, tikagrelor.
- jeśli pacjent przyjmuje leki przeciwzapalne, takie jak diklofenak, ibuprofen, piroksykam.
- jeśli u pacjenta występuje infekcja serca (bakteryjne zapalenie wsierdzia).
- jeśli pacjent wie, że czynność nerek jest zaburzona lub pacjent jest odwodniony (objawy obejmują uczucie pragnienia i oddawanie moczu w zmniejszonej ilości ciemnego (skoncentrowanego) moczu).
- jeśli masz ponad 75 lat.
- jeśli waży 50 kg lub mniej.
- jeśli miałeś zawał serca lub zdiagnozowano u Ciebie schorzenia zwiększające ryzyko wystąpienia zawału serca.
- jeśli przechodzisz planowaną operację. Pradaxa będzie musiała zostać tymczasowo odstawiona ze względu na zwiększone ryzyko krwawienia w trakcie i wkrótce po operacji. Jeśli to możliwe, lek Pradaxa należy odstawić co najmniej 24 godziny przed operacją.U pacjentów ze zwiększonym ryzykiem krwawienia lekarz może podjąć decyzję o wcześniejszym przerwaniu leczenia.
- jeśli przechodzisz nieplanowany zabieg chirurgiczny. Jeśli to możliwe, zabieg chirurgiczny należy odroczyć do 12 godzin po podaniu ostatniej dawki leku Pradaxa. Jeśli nie można odroczyć operacji, może wystąpić zwiększone ryzyko krwawienia. Lekarz oceni ryzyko krwawienia i pilność operacji.
- jeśli masz rurkę (cewnik) założoną w plecy: rurkę można wprowadzić w plecy, np. w celu podawania środków znieczulających lub przeciwbólowych podczas zabiegu chirurgicznego lub po nim.Jeśli pacjent otrzyma lek Pradaxa po usunięciu cewnika, lekarz będzie regularnie kontrolował stan pacjenta.
- jeśli upadniesz lub zranisz się podczas leczenia, zwłaszcza jeśli dostaniesz cios w głowę, natychmiast skontaktuj się z lekarzem. Twój lekarz może uznać za konieczne spotkanie z tobą, ponieważ możesz być narażony na wysokie ryzyko krwawienia.
Dzieci i młodzież
Leku Pradaxa nie należy stosować u dzieci i młodzieży w wieku poniżej 18 lat.
Interakcje Jakie leki lub pokarmy mogą zmienić działanie leku Pradaxa
Należy powiedzieć lekarzowi lub farmaceucie, jeśli pacjent przyjmuje, ostatnio przyjmował lub może przyjmować jakiekolwiek inne leki. Np:
- Leki zmniejszające krzepliwość krwi (np. warfaryna, fenprokumon, heparyna, klopidogrel, prasugrel, tikagrelor, rywaroksaban)
- Leki przeciwzapalne i przeciwbólowe (np. aspiryna)
- Ziele dziurawca, lek ziołowy w leczeniu depresji
- Leki przeciwdepresyjne zwane selektywnymi inhibitorami wychwytu zwrotnego serotoniny lub selektywnymi inhibitorami wychwytu zwrotnego serotoniny i noradrenaliny
- Ryfampicyna lub klarytromycyna, dwa antybiotyki
- Leki stosowane w leczeniu zaburzeń rytmu serca (np. amiodaron, dronedaron, chinidyna, werapamil). Jeśli pacjent przyjmuje leki zawierające werapamil, powinien przyjąć zmniejszoną dawkę leku Pradaxa, wynoszącą 220 mg, przyjmowaną w postaci jednej kapsułki 110 mg dwa razy na dobę, ponieważ może zwiększyć się ryzyko krwawienia.Pradaxa i leki zawierające werapamil należy przyjmować jednocześnie.
- Leki stosowane w leczeniu zakażeń grzybiczych (np. ketokonazol, itrakonazol, pozakonazol), chyba że są stosowane wyłącznie na skórę
- Leki zapobiegające bezpośrednim epizodom po przeszczepieniu narządu (np. takrolimus, cyklosporyna)
- Leki wirusowe na AIDS (np. rytonawir)
- Leki stosowane w leczeniu padaczki (np. karbamazepina, fenytoina)
Ostrzeżenia Ważne jest, aby wiedzieć, że:
Ciąża i karmienie piersią
Wpływ leku Pradaxa na ciążę i płód nie jest znany. Nie należy przyjmować leku Pradaxa, jeśli pacjentka jest w ciąży, chyba że lekarz stwierdzi, że jest to bezpieczne. Kobiety w wieku rozrodczym muszą unikać zajścia w ciążę podczas leczenia lekiem Pradaxa.
Nie należy karmić piersią podczas leczenia lekiem Pradaxa.
Prowadzenie i używanie maszyn
Pradaxa nie ma znanego wpływu na zdolność prowadzenia pojazdów i obsługiwania maszyn.
Pradaxa zawiera żółcień pomarańczową (E110)
Ten lek zawiera barwnik zwany żółcień pomarańczową (E110), który może powodować reakcje alergiczne
Dawka, sposób i czas podawania Jak stosować lek Pradaxa: dawkowanie
Lek należy zawsze stosować zgodnie z zaleceniami lekarza. W razie wątpliwości skonsultuj się z lekarzem.
Lek Pradaxa należy przyjmować zgodnie z zaleceniami w następujących stanach:
Zapobieganie powstawaniu zakrzepów krwi po operacji wymiany stawu kolanowego lub biodrowego
Zalecana dawka to 300 mg raz na dobę (przyjmowana jako 2 kapsułki po 150 mg).
Jeśli czynność nerek jest zmniejszona o więcej niż połowę lub jeśli masz 75 lat lub więcej, zalecana dawka to 220 mg przyjmowana jako jedna kapsułka 110 mg dwa razy dziennie.
Jeśli pacjent przyjmuje leki zawierające werapamil, powinien przyjmować zmniejszoną dawkę leku Pradaxa, wynoszącą 220 mg, przyjmowaną w postaci jednej kapsułki 110 mg dwa razy na dobę, ponieważ ryzyko krwawienia może się zwiększyć.
Jeśli u pacjenta występuje zwiększone ryzyko krwawienia, lekarz może zdecydować o przepisaniu dawki 220 mg leku Pradaxa przyjmowanej w postaci jednej kapsułki 110 mg dwa razy na dobę.
Lek Pradaxa można przyjmować z jedzeniem lub bez jedzenia. Kapsułkę należy połykać w całości, popijając szklanką wody, aby zapewnić uwolnienie z żołądka.Nie należy łamać, żuć ani wyjmować granulek z kapsułki, ponieważ może to zwiększyć ryzyko krwawienia.
W przypadku stosowania leku Pradaxa w blistrze należy przestrzegać następujących instrukcji
- wyjąć kapsułki z blistra, unosząc folię aluminiową z tyłu.
- nie wypychać kapsułek przez blister.
- folię aluminiową z blistra należy unieść tylko wtedy, gdy ma zostać wyjęta kapsułka.
W przypadku stosowania leku Pradaxa zapakowanego w butelkę należy przestrzegać następujących instrukcji
- butelkę otwiera się przez naciśnięcie i przekręcenie zakrętki.
Zmiana w leczeniu przeciwzakrzepowym
- Zmiana leczenia lekiem Pradaxa na leczenie lekami przeciwzakrzepowymi podawanymi we wstrzyknięciach: Nie należy rozpoczynać leczenia lekami przeciwzakrzepowymi do wstrzykiwań (np. heparyną) przed upływem 12 godzin od ostatniego podania leku Pradaxa.
- Zmiana z leczenia lekami przeciwzakrzepowymi podawanymi we wstrzyknięciu na leczenie lekiem Pradaxa: Rozpocznij przyjmowanie leku Pradaxa 0-2 godziny przed następnym wstrzyknięciem.
- Zmiana z leczenia lekiem Pradaxa na leczenie lekami rozrzedzającymi krew zawierającymi antagonistów witaminy K (np. fenprokumon): lekarz powinien zlecić wykonanie badań krwi i powiedzieć, kiedy należy rozpocząć leczenie antagonistami witaminy K.
- Zmiana leczenia lekami rozrzedzającymi krew zawierającymi antagonistów witaminy K (np. fenprokumon) na leczenie lekiem Pradaxa: Należy przerwać przyjmowanie leku zawierającego antagonistę witaminy K. Lekarz powinien zlecić wykonanie badań krwi i poinformować o rozpoczęciu leczenia lekiem Pradaxa.
Przedawkowanie Co zrobić w przypadku przyjęcia zbyt dużej dawki leku Pradaxa
Przyjęcie większej niż zalecana dawki leku Pradaxa
W przypadku przyjęcia większej dawki leku Pradaxa niż zalecana, może wystąpić zwiększone ryzyko krwawienia. Twój lekarz może wykonać badanie krwi, aby ocenić ryzyko krwawienia.
W przypadku przyjęcia większej dawki leku Pradaxa niż przepisana, należy natychmiast poinformować lekarza. Jeśli wystąpi epizod krwawienia, może być wymagany zabieg chirurgiczny lub leczenie za pomocą transfuzji krwi.
Pominięcie przyjęcia leku Pradaxa
Pominiętą dawkę można przyjąć do 6 godzin przed kolejną dawką.Jeśli do następnej dawki pozostało mniej niż 6 godzin, pominiętą dawkę należy pominąć. Nie należy stosować dawki podwójnej w celu uzupełnienia pominiętej dawki
Przerwanie stosowania leku Pradaxa
Lek Pradaxa należy przyjmować dokładnie zgodnie z zaleceniami. Nie należy przerywać stosowania leku Pradaxa bez uprzedniej konsultacji z lekarzem.Przerwanie stosowania leku Pradaxa może zwiększyć ryzyko powstania zakrzepów krwi u pacjentów leczonych po operacji wymiany stawu biodrowego lub kolanowego.
W przypadku dalszych pytań dotyczących stosowania tego leku należy zwrócić się do lekarza lub farmaceuty.
Skutki uboczne Jakie są skutki uboczne Pradaxa
Jak każdy lek, lek ten może powodować działania niepożądane, chociaż nie u każdego one wystąpią.
Pradaxa działa na układ krzepnięcia krwi, więc większość działań niepożądanych jest związana z objawami, takimi jak krwiak lub krwawienie.
Mogą wystąpić poważne lub ciężkie krwawienia, są to najpoważniejsze skutki uboczne, które niezależnie od lokalizacji mogą powodować kalectwo, zagrażać życiu, a nawet prowadzić do śmierci. W niektórych przypadkach te krwawienia mogą nie być widoczne.
Jeśli wystąpi jakiekolwiek krwawienie, które nie ustępuje samoistnie lub jeśli wystąpią objawy nadmiernego krwawienia (wyjątkowe osłabienie, uczucie zmęczenia, bladość skóry, zawroty głowy, ból głowy lub niewyjaśniony obrzęk), należy natychmiast skontaktować się z lekarzem.
Lekarz może zdecydować o dokładnym zbadaniu pacjenta lub o zmianie leczenia.
Należy natychmiast poinformować lekarza, jeśli wystąpi ciężka reakcja alergiczna, która powoduje trudności w oddychaniu lub zawroty głowy.
Skutki uboczne są wymienione poniżej, pogrupowane według częstotliwości ich występowania.
Zapobieganie zablokowaniu naczyń krwionośnych w mózgu lub reszcie ciała z powodu tworzenia się skrzepów krwi, które powstały w wyniku zmienionego bicia serca
Często (mogą dotyczyć do 1 na 10 osób):
- Krwawienie, które może wystąpić z nosa, żołądka lub jelit, prącia/pochwy lub dróg moczowych (w tym krew w moczu, która sprawia, że jest różowa lub czerwona) lub pod skórą
- Zmniejszenie liczby czerwonych krwinek we krwi
- Ból brzucha lub żołądka
- Niestrawność
- Biegunka ze słabo uformowanymi lub płynnymi stolcami
- Czuję się niedobrze
Niezbyt często (może dotyczyć nie więcej niż 1 na 100 osób):
- Krwawienie
- Krwawienie, które może wystąpić z hemoroidów, do odbytnicy lub mózgu
- Powstawanie krwiaka
- Odkrztuszanie krwi lub plwociny w kolorze krwi
- Zmniejszenie liczby płytek krwi
- Zmniejszenie ilości hemoglobiny we krwi (substancja znajdująca się w czerwonych krwinkach)
- Reakcja alergiczna
- Nagła zmiana skóry, która zmienia jej kolor i wygląd
- Swędzący
- Wrzód żołądkowo-jelitowy (w tym wrzód przełyku)
- Zapalenie przełyku i żołądka
- Refluks soków żołądkowych w przełyku
- On wymiotował
- Trudności z połykaniem
- Nieprawidłowe wyniki testów czynności wątroby
Rzadko (może dotyczyć do 1 na 1000 osób):
- Krwawienie, które może wystąpić w stawie, z nacięcia chirurgicznego, z rany, z miejsca wstrzyknięcia lub z miejsca wprowadzenia cewnika do żyły - Ciężka reakcja alergiczna powodująca trudności w oddychaniu lub zawroty głowy - Ciężka reakcja alergiczna powodująca obrzęk twarzy lub wysypka skórna widoczna w postaci ciemnoczerwonych, opuchniętych, swędzących grudek wywołanych reakcją alergiczną - zmniejszenie liczby czerwonych krwinek - zwiększenie aktywności enzymów wątrobowych - zażółcenie skóry lub białkówek oczu z powodu problemów z wątrobą lub krwią
Nieznana (częstość nie może być określona na podstawie dostępnych danych):
- Trudności w oddychaniu lub świszczący oddech
Leczenie zakrzepów krwi w żyłach nóg i płuc, w tym zapobieganie ponownemu tworzeniu się zakrzepów krwi w żyłach nóg i (lub) płucach
Często (mogą dotyczyć do 1 na 10 osób):
- Krwawienie, które może wystąpić z nosa, żołądka lub jelit, odbytnicy, penisa/pochwy lub dróg moczowych (w tym krew w moczu, która sprawia, że jest różowa lub czerwona) lub pod skórą
- Niestrawność
Niezbyt często (może dotyczyć nie więcej niż 1 na 100 osób):
- Krwawienie
- Krwawienie, które może wystąpić w stawie z rany
- Krwawienie, które może wystąpić z hemoroidów
- Zmniejszenie liczby czerwonych krwinek we krwi
- Powstawanie krwiaka
- Odkrztuszanie krwi lub zakrwawionej plwociny
- Reakcja alergiczna
- Nagła zmiana skóry, która zmienia jej kolor i wygląd
- Swędzący
- Wrzód żołądkowo-jelitowy
- Zapalenie przełyku i żołądka
- Refluks soków żołądkowych w przełyku
- Czuję się niedobrze
- On wymiotował
- Ból brzucha lub żołądka
- Biegunka ze słabo uformowanymi lub płynnymi stolcami
- Trudności z połykaniem
- Nieprawidłowe wyniki testów czynności wątroby
- Zwiększone enzymy wątrobowe
Rzadko (może dotyczyć do 1 na 1000 osób):
- Krwawienie, które może wystąpić z „nacięcia chirurgicznego lub z miejsca wstrzyknięcia lub z miejsca wprowadzenia cewnika do żyły lub z mózgu
- Zmniejszenie liczby płytek krwi
- Ciężka reakcja alergiczna powodująca trudności w oddychaniu lub zawroty głowy
- Ciężka reakcja alergiczna powodująca obrzęk twarzy lub gardła
- Zauważalna wysypka skórna z ciemnoczerwonymi, opuchniętymi, swędzącymi grudkami spowodowanymi reakcją alergiczną
- Trudności z połykaniem
- Zmniejszenie liczby czerwonych krwinek we krwi
Nieznana (częstość nie może być określona na podstawie dostępnych danych):
- Trudności w oddychaniu lub świszczący oddech
- Zmniejszenie ilości hemoglobiny obecnej we krwi (substancja zawarta w czerwonych krwinkach)
- Zmniejszenie liczby czerwonych krwinek we krwi
- Zażółcenie skóry lub białkówek oczu spowodowane problemami z wątrobą lub krwią
Zgłaszanie skutków ubocznych
W przypadku wystąpienia jakichkolwiek działań niepożądanych należy zwrócić się do lekarza lub farmaceuty, w tym działań niepożądanych niewymienionych w tej ulotce.Można również zgłaszać działania niepożądane bezpośrednio za pośrednictwem krajowego systemu zgłaszania.Więcej informacji na temat bezpieczeństwa stosowania tego leku.
W badaniu klinicznym częstość zawałów serca była wyższa w przypadku leku Pradaxa niż warfaryny. Ogólna częstość występowania była niska.
Wygaśnięcie i przechowywanie
Lek należy przechowywać w miejscu niewidocznym i niedostępnym dla dzieci.
Nie stosować tego leku po upływie terminu ważności zamieszczonego na pudełku, blistrze lub butelce po EXP.Termin ważności oznacza ostatni dzień podanego miesiąca.
Blistry: Przechowywać w oryginalnym opakowaniu w celu ochrony przed wilgocią.
Butelka: Po otwarciu lek należy zużyć w ciągu 4 miesięcy. Trzymaj butelkę szczelnie zamkniętą. Przechowywać w oryginalnym opakowaniu w celu ochrony przed wilgocią.
Nie należy wyrzucać żadnych leków do kanalizacji ani domowych pojemników na odpadki. Należy zapytać farmaceutę, jak usunąć leki, których się już nie używa.Pomoże to chronić środowisko.
Skład i postać farmaceutyczna
Co zawiera lek Pradaxa
- Substancją czynną jest dabigatran, który podawany jest w postaci eteksylanu dabigatranu jako mesylan eteksylanu dabigatranu w dawce 150mg
- Pozostałe składniki to kwas winowy, guma arabska, hypromeloza, dimetikon 350, talk i hydroksypropyloceluloza.
- Otoczka kapsułki zawiera karageninę, chlorek potasu, dwutlenek tytanu, indygokarmin, żółcień pomarańczową (E110), hypromelozę i wodę oczyszczoną.
- Czarny tusz drukarski zawiera szelak, alkohol N-butylowy, alkohol izopropylowy, przemysłowy denaturowany etanol, czarny tlenek żelaza, wodę oczyszczoną i glikol propylenowy.
Opis wyglądu leku Pradaxa i co zawiera opakowanie
Pradaxa to kapsułka twarda.
Kapsułki twarde Pradaxa 150 mg mają nieprzezroczyste jasnoniebieskie wieczko i nieprzezroczysty kremowy korpus. Na wieczku nadrukowano logo Boehringer Ingelheim, a na korpusie kapsułki kod „R150”.
Pradaxa 150 mg kapsułki twarde są dostępne w opakowaniach zawierających 10x1, 30x1, 60x1 kapsułek twardych, w opakowaniu zbiorczym zawierającym 3 opakowania po 60x1 kapsułek twardych (180 kapsułek twardych) lub w opakowaniu zbiorczym zawierającym 2 opakowania po 50x1 kapsułek twardych (100 kapsułek twardych) w aluminium blistry podzielne według dawki jednostkowej.
Pradaxa 150 mg kapsułki twarde są również dostępne w opakowaniach zawierających 60x1 kapsułek twardych w białych perforowanych blistrach aluminiowych podzielonych na dawki pojedyncze.
Kapsułki twarde Pradaxa 150 mg są również dostępne w butelkach polipropylenowych (plastikowych) zawierających 60 kapsułek twardych.
Nie wszystkie rozmiary opakowań mogą być wprowadzone na rynek
Ulotka pakietu źródłowego: AIFA (Włoska Agencja Leków). Treść opublikowana w styczniu 2016 r. Przedstawione informacje mogą być nieaktualne.
Aby mieć dostęp do najbardziej aktualnej wersji, warto wejść na stronę AIFA (Włoskiej Agencji Leków). Zastrzeżenie i przydatne informacje.
01.0 NAZWA PRODUKTU LECZNICZEGO
PRADAXA 110 MG KAPSUŁEK TWARDYCH
02.0 SKŁAD JAKOŚCIOWY I ILOŚCIOWY
Każda kapsułka twarda zawiera 110 mg eteksylanu dabigatranu (w postaci mesylanu).
Substancje pomocnicze o znanym działaniu:
Każda kapsułka twarda zawiera 3 mikrogramy żółcieni pomarańczowej (E110).
Pełny wykaz substancji pomocniczych, patrz punkt 6.1.
03.0 POSTAĆ FARMACEUTYCZNA
Kapsułka twarda.
Kapsułki z nieprzezroczystym jasnoniebieskim wieczkiem i nieprzezroczystym kremowym korpusem w rozmiarze 1 wypełnione żółtawymi peletkami. Logo Boehringer Ingelheim nadrukowane na głowie, „R110” na korpusie.
04.0 INFORMACJE KLINICZNE
04.1 Wskazania terapeutyczne
Profilaktyka pierwotna epizodów zakrzepowo-zatorowych u dorosłych pacjentów poddawanych planowej całkowitej alloplastyce stawu biodrowego lub kolanowego.
Zapobieganie udarom mózgu i zatorowości systemowej u dorosłych pacjentów z niezastawkowym migotaniem przedsionków (NVAF), z jednym lub większą liczbą czynników ryzyka, takich jak przebyty udar lub przemijający napad niedokrwienny (TIA); wiek ≥ 75 lat; niewydolność serca (klasa NYHA ≥ II); cukrzyca; nadciśnienie.
Leczenie zakrzepicy żył głębokich (ZŻG) i zatorowości płucnej (ZP) oraz zapobieganie nawrotom ZŻG i ZP u dorosłych.
04.2 Dawkowanie i sposób podawania
Dawkowanie
Prewencja pierwotna żylnych epizodów zakrzepowo-zatorowych w chirurgii ortopedycznej
Pacjenci poddawani planowej operacji wymiany stawu kolanowego
Zalecana dawka leku Pradaxa to 220 mg raz na dobę, przyjmowana jako 2 kapsułki po 110 mg. Leczenie należy rozpocząć doustnie w ciągu 1-4 godzin po zakończeniu zabiegu od jednej kapsułki 110 mg i kontynuować od następnego dnia podając 2 kapsułki raz dziennie przez łącznie 10 dni.
Pacjenci poddawani planowej operacji wymiany stawu biodrowego
Zalecana dawka leku Pradaxa to 220 mg raz na dobę, przyjmowana jako 2 kapsułki po 110 mg. Leczenie należy rozpocząć doustnie w ciągu 1-4 godzin po zakończeniu zabiegu, stosując kapsułkę 110 mg i kontynuować od następnego dnia podając 2 kapsułki raz na dobę, łącznie przez 28-35 dni.
Dla poniższych grup zalecana dawka dobowa leku Pradaxa wynosi 150 mg raz na dobę, przyjmowana jako 2 kapsułki po 75 mg.
Leczenie należy rozpocząć doustnie w ciągu 1-4 godzin po zakończeniu zabiegu jedną kapsułką 75 mg i kontynuować od następnego dnia podając 2 kapsułki raz na dobę przez łącznie 10 dni (operacja stawu kolanowego) lub 28 dni -35 dni (wymiana stawu biodrowego Chirurgia):
• Pacjenci z umiarkowanymi zaburzeniami czynności nerek (klirens kreatyniny, CrCL 30-50 ml/min [patrz Zaburzenie czynności nerek (pierwotna profilaktyka żylnych epizodów zakrzepowo-zatorowych w chirurgii ortopedycznej)]
• Pacjenci jednocześnie otrzymujący werapamil, amiodaron, chinidynę [patrz Jednoczesne stosowanie produktu Pradaxa ze słabymi lub umiarkowanymi inhibitorami glikoproteiny P (P-gp), takimi jak amiodaron, chinidyna lub werapamil (pierwotna profilaktyka żylnej choroby zakrzepowo-zatorowej w chirurgii ortopedycznej)]
• Pacjenci w wieku 75 lat i starsi [patrz Pacjenci w podeszłym wieku (pierwotna profilaktyka żylnej choroby zakrzepowo-zatorowej w chirurgii ortopedycznej)]
W przypadku obu interwencji, jeśli hemostaza nie jest prawidłowa, rozpoczęcie leczenia należy odroczyć. Jeżeli leczenie nie zostanie rozpoczęte w dniu zabiegu, należy rozpocząć od 2 kapsułek raz dziennie.
Ocena czynności nerek (prewencja pierwotna epizodów żylnej choroby zakrzepowo-zatorowej w chirurgii ortopedycznej):
U wszystkich pacjentów:
• Czynność nerek należy ocenić, obliczając klirens kreatyniny (CrCL) przed rozpoczęciem leczenia produktem Pradaxa, aby wykluczyć pacjentów z ciężkimi zaburzeniami czynności nerek (tj. CrCL).
• Czynność nerek należy również ocenić, gdy podejrzewa się pogorszenie czynności nerek podczas leczenia (np. hipowolemia, odwodnienie oraz w przypadku jednoczesnego stosowania niektórych produktów leczniczych).
Metodą stosowaną do oceny czynności nerek (CrCL w ml/min) podczas klinicznego rozwoju produktu Pradaxa była metoda Cockgroft-Gault (patrz punkt 4.2 produktu Pradaxa 75 mg).
Populacje specjalne
Zaburzenia czynności nerek (pierwotna prewencja epizodów żylnej choroby zakrzepowo-zatorowej w chirurgii ortopedycznej)
Leczenie produktem Pradaxa u pacjentów z ciężkimi zaburzeniami czynności nerek (CrCLr
Doświadczenie kliniczne u pacjentów z umiarkowanymi zaburzeniami czynności nerek (CrCL 30-50 ml/min) jest ograniczone.Tych pacjentów należy leczyć ostrożnie.Zalecana dawka to 150 mg przyjmowana raz na dobę jako 2 kapsułki po 75 mg (patrz punkty 4.4 i 5.1).
Jednoczesne stosowanie produktu Pradaxa ze słabymi lub umiarkowanymi inhibitorami glikoproteiny P (P-gp), takimi jak amiodaron, chinidyna lub werapamil (pierwotna profilaktyka żylnej choroby zakrzepowo-zatorowej w chirurgii ortopedycznej)
U pacjentów otrzymujących jednocześnie eteksylan dabigatranu i amiodaron, chinidynę lub werapamil, dawkę produktu Pradaxa należy zmniejszyć do 150 mg przyjmowanych raz na dobę w postaci dwóch kapsułek 75 mg (patrz punkty 4.4 i 4.5). W takim przypadku lek Pradaxa i te leki należy przyjmować razem.
U pacjentów z umiarkowanymi zaburzeniami czynności nerek, którzy są jednocześnie leczeni eteksylanem dabigatranu i werapamilem, należy rozważyć zmniejszenie dawki produktu Pradaxa do 75 mg na dobę (patrz punkty 4.4 i 4.5).
Osoby w podeszłym wieku (pierwotna profilaktyka epizodów żylnej choroby zakrzepowo-zatorowej w chirurgii ortopedycznej)
Doświadczenie kliniczne u pacjentów w podeszłym wieku (>75 lat) jest ograniczone.Tych pacjentów należy leczyć ostrożnie.Zalecana dawka to 150 mg przyjmowane raz na dobę w postaci dwóch kapsułek po 75 mg (patrz punkty 4.4 i 5.1).
Ponieważ zaburzenia czynności nerek mogą występować często u osób w podeszłym wieku (wiek > 75 lat), czynność nerek należy ocenić, obliczając CrCL przed rozpoczęciem leczenia produktem Pradaxa, aby wykluczyć pacjentów z ciężkimi zaburzeniami czynności nerek (tj.
Zaburzenia czynności wątroby (pierwotna prewencja epizodów żylnej choroby zakrzepowo-zatorowej w chirurgii ortopedycznej)
Pacjenci z aktywnością enzymów wątrobowych dwukrotnie przekraczającą górną granicę normy (GGN) byli wykluczeni z badań klinicznych oceniających zapobieganie VTE po planowej alloplastyce stawu biodrowego lub kolanowego. stosowanie produktu Pradaxa nie jest zalecane w tej populacji (patrz punkty 4.4 i 5.2).Jest przeciwwskazany w przypadku niewydolności wątroby lub choroby wątroby, która może mieć wpływ na przeżycie (patrz punkt 4.3).
Waga (pierwotna profilaktyka żylnych epizodów zakrzepowo-zatorowych w chirurgii ortopedycznej)
Doświadczenie kliniczne z zalecaną dawką u pacjentów o masie ciała 110 kg jest bardzo ograniczone.Na podstawie danych klinicznych i kinetycznych nie jest konieczne dostosowanie dawki (patrz punkt 5.2), ale zaleca się ścisłą obserwację kliniczną (patrz punkt 4.4).
Płeć (pierwotna prewencja epizodów żylnej choroby zakrzepowo-zatorowej w chirurgii ortopedycznej)
Na podstawie dostępnych danych klinicznych i kinetycznych nie jest wymagane dostosowanie dawki (patrz punkt 5.2).
Switching (prewencja pierwotna epizodów żylnej choroby zakrzepowo-zatorowej w chirurgii ortopedycznej)
Od leczenia lekiem Pradaxa do pozajelitowego leku przeciwzakrzepowego
Zaleca się odczekanie 24 godzin po przyjęciu ostatniej dawki przed zmianą leku Pradaxa na pozajelitowy lek przeciwzakrzepowy (patrz punkt 4.5).
Od pozajelitowych leków przeciwzakrzepowych po Pradaxa
Wstrzymać pozajelitowy lek przeciwzakrzepowy i rozpocząć leczenie eteksylanem dabigatranu na 0-2 godziny przed planowanym podaniem kolejnej dawki pierwotnego leczenia lub po przerwaniu leczenia w przypadku ciągłego leczenia (np. dożylna heparyna niefrakcjonowana (ENF)) (patrz punkt 4.5).
Populacja pediatryczna (prewencja pierwotna epizodów żylnej choroby zakrzepowo-zatorowej w chirurgii ortopedycznej)
Stosowanie produktu Pradaxa u dzieci i młodzieży nie jest właściwe we wskazaniu: pierwotna prewencja epizodów żylnej choroby zakrzepowo-zatorowej u pacjentów poddawanych planowej alloplastyce całkowitej stawu biodrowego lub planowej całkowitej alloplastyce stawu kolanowego.
Pominięta dawka (pierwotna prewencja epizodów żylnej choroby zakrzepowo-zatorowej w chirurgii ortopedycznej)
Zaleca się kontynuację pozostałych dawek dobowych eteksylanu dabigatranu o tej samej porze następnego dnia.
Nie należy podwajać dawek w celu uzupełnienia pominiętej dawki.
Dawkowanie (profilaktyka udaru mózgu u pacjentów z migotaniem przedsionków, ZŻG/ZP)
Profilaktyka udaru mózgu i ES u dorosłych pacjentów z NLPZ z jednym lub kilkoma czynnikami ryzyka (profilaktyka udaru mózgu u pacjentów z migotaniem przedsionków)
Zalecana dawka dobowa leku Pradaxa to 300 mg przyjmowana jako jedna kapsułka 150 mg dwa razy na dobę. Terapia musi być kontynuowana długoterminowo.
Leczenie zakrzepicy żył głębokich (ZŻG) i zatorowości płucnej (ZP) oraz zapobieganie nawrotom ZŻG i ZP u dorosłych (ZŻG/ZP)
Zalecana dawka dobowa leku Pradaxa to 300 mg przyjmowana w postaci jednej kapsułki 150 mg dwa razy na dobę, po leczeniu pozajelitowym lekiem przeciwzakrzepowym podawanym przez co najmniej 5 dni. Czas trwania leczenia należy ustalić po „dokładnej ocenie korzyści z leczenia w porównaniu z ryzykiem krwawienia (patrz punkt 4.4). Wybór terapii krótkotrwałej (co najmniej 3 miesiące) powinien opierać się na przemijających czynnikach ryzyka (np. niedawno przebyty zabieg chirurgiczny, uraz, unieruchomienie) oraz te, które trwają dłużej z powodu stałych czynników ryzyka lub idiopatycznej ZŻG lub ZP.
Profilaktyka udaru mózgu u pacjentów z migotaniem przedsionków, ZŻG/ZP
Dla następujących grup zalecana dzienna dawka leku Pradaxa wynosi 220 mg w postaci jednej kapsułki 110 mg dwa razy dziennie:
• Pacjenci w wieku 80 lat lub starsi
• Pacjenci leczeni jednocześnie werapamilem
W następujących grupach dawka dobowa produktu Pradaxa 300 mg lub 220 mg powinna być ustalana indywidualnie na podstawie oceny ryzyka zakrzepowo-zatorowego i ryzyka krwawienia:
• Pacjenci w wieku od 75 do 80 lat
• Pacjenci z umiarkowanymi zaburzeniami czynności nerek
• Pacjenci z zapaleniem żołądka, zapaleniem przełyku lub refluksem żołądkowo-przełykowym
• Inni pacjenci ze zwiększonym ryzykiem krwawienia
W przypadku ZŻG/ZP zalecenie stosowania produktu Pradaxa w dawce 220 mg przyjmowanego w postaci jednej kapsułki 110 mg dwa razy na dobę jest oparte na ocenach farmakokinetycznych i farmakodynamicznych i nie było badane w tej podgrupie klinicznej.
Więcej informacji można znaleźć w sekcjach 4.4, 4.5, 5.1 i 5.2 poniżej.
W przypadku nietolerancji dabigatranu należy poinstruować pacjentów, aby natychmiast skontaktowali się z lekarzem, który może przenieść ich na akceptowalne alternatywne opcje leczenia w celu zapobiegania udarowi i ES związanemu z migotaniem przedsionków lub ZŻG/ZP.
Osoby w podeszłym wieku (profilaktyka udarów u pacjentów z migotaniem przedsionków, ZŻG/ZP)
Pacjenci w wieku od 75 do 80 lat powinni być leczeni dawką dobową 300 mg przyjmowaną jako jedna kapsułka 150 mg dwa razy na dobę. W indywidualnych przypadkach i według uznania lekarza można rozważyć dawkę dobową 220 mg przyjmowaną jako jedna kapsułka 110 mg dwa razy na dobę, gdy ryzyko zakrzepicy zatorowej jest niskie, a ryzyko krwawienia wysokie (patrz punkt 4.4).
Pacjenci w wieku 80 lat lub starsi powinni być leczeni dawką dobową 220 mg przyjmowaną jako jedna kapsułka 110 mg dwa razy na dobę, ze względu na zwiększone ryzyko krwawienia w tej populacji.
Ponieważ zaburzenia czynności nerek mogą występować często u osób w podeszłym wieku (wiek > 75 lat), czynność nerek należy ocenić, obliczając CrCL przed rozpoczęciem leczenia produktem Pradaxa, aby wykluczyć pacjentów z ciężkimi zaburzeniami czynności nerek (tj.
Pacjenci zagrożeni krwawieniem (profilaktyka udarów u pacjentów z migotaniem przedsionków, ZŻG/ZP)
Pacjenci ze zwiększonym ryzykiem krwawienia (patrz punkty 4.4, 4.5, 5.1 i 5.2) powinni być poddani „dokładnej obserwacji klinicznej (w poszukiwaniu oznak krwawienia lub niedokrwistości). korzyści i potencjalne ryzyko dla każdego indywidualnego pacjenta. Test krzepnięcia (patrz punkt 4.4) może pomóc w identyfikacji pacjentów ze zwiększonym ryzykiem krwawienia z powodu „zwiększonej ekspozycji na dabigatran. W przypadku stwierdzenia nadmiernej ekspozycji na dabigatran u pacjentów z wysokim ryzykiem krwawienia, dawka 220 mg przyjmowana jako jedna 110 kapsułka mg dwa razy na dobę. W przypadku wystąpienia klinicznie istotnego krwawienia leczenie należy przerwać.
U osób z zapaleniem błony śluzowej żołądka, zapaleniem przełyku lub refluksem przełyku można rozważyć dawkę 220 mg przyjmowaną jako jedna kapsułka 110 mg dwa razy na dobę ze względu na wysokie ryzyko wystąpienia poważnych krwawień z przewodu pokarmowego (patrz punkt 4.4).
Ocena czynności nerek (profilaktyka udaru mózgu u pacjentów z migotaniem przedsionków, ZŻG/ZP):
U wszystkich pacjentów:
• Czynność nerek należy ocenić, obliczając klirens kreatyniny (CrCL) przed rozpoczęciem leczenia produktem Pradaxa, aby wykluczyć pacjentów z ciężkimi zaburzeniami czynności nerek (tj. CrCL).
• Czynność nerek należy również ocenić, gdy podejrzewa się pogorszenie czynności nerek podczas leczenia (np. hipowolemia, odwodnienie oraz w przypadku jednoczesnego stosowania niektórych produktów leczniczych).
Dodatkowe wymagania dla pacjentów z łagodnymi do umiarkowanych zaburzeniami czynności nerek oraz dla pacjentów w wieku powyżej 75 lat:
• Czynność nerek należy oceniać podczas leczenia produktem Pradaxa co najmniej raz w roku lub częściej, jeśli jest to wymagane w pewnych sytuacjach klinicznych, gdy podejrzewa się pogorszenie lub pogorszenie czynności nerek (np. hipowolemia, odwodnienie oraz w przypadku jednoczesnego stosowania niektórych leków).
Metodą stosowaną do oceny czynności nerek (CrCL w ml/min) podczas klinicznego rozwoju produktu Pradaxa była metoda Cockgroft-Gault (patrz punkt 4.2 produktu Pradaxa 75 mg).
Zaburzenia czynności nerek (profilaktyka udaru mózgu u pacjentów z migotaniem przedsionków, ZŻG/ZP)
Leczenie preparatem Pradaxa u pacjentów z ciężkimi zaburzeniami czynności nerek (CrCLr
Nie ma konieczności dostosowania dawki u pacjentów z łagodnymi zaburzeniami czynności nerek (CrCL 50-≤ 80 ml/min). Nawet u pacjentów z umiarkowanymi zaburzeniami czynności nerek (CrCL 30-50 ml/min) zalecana dawka leku Pradaxa wynosi 300 mg przyjmowana jako jedna kapsułka 150 mg dwa razy na dobę. Jednak u pacjentów z wysokim ryzykiem krwawienia należy rozważyć zmniejszenie dawki produktu Pradaxa do 220 mg przyjmowanych w postaci jednej kapsułki 110 mg dwa razy na dobę (patrz punkty 4.4 i 5.2). Zaleca się ścisłą obserwację kliniczną pacjentów z zaburzeniami czynności nerek.
Jednoczesne stosowanie leku Pradaxa ze słabymi lub umiarkowanymi inhibitorami glikoproteiny P (P-gp), takimi jak amiodaron, chinidyna lub werapamil (zapobieganie udarowi mózgu u pacjentów z migotaniem przedsionków, ZŻG/ZP)
Nie ma konieczności dostosowania dawki w przypadku jednoczesnego stosowania z amiodaronem lub chinidyną (patrz punkty 4.4, 4.5 i 5.2).
U pacjentów otrzymujących zarówno eteksylan dabigatranu, jak i werapamil, dawkę dabigatranu należy zmniejszyć do 220 mg przyjmowanych w postaci jednej kapsułki 110 mg dwa razy na dobę (patrz punkty 4.4 i 4.5). W takim przypadku Pradaxa i werapamil muszą być przyjmowane razem.
Waga (profilaktyka udarów u pacjentów z migotaniem przedsionków, ZŻG/ZP)
Na podstawie dostępnych danych klinicznych i kinetycznych nie jest konieczne dostosowanie dawki (patrz punkt 5.2), ale zaleca się ścisłą obserwację kliniczną pacjentów z masą ciała.
Płeć (profilaktyka udaru mózgu u pacjentów z migotaniem przedsionków, ZŻG/ZP)
Na podstawie dostępnych danych klinicznych i kinetycznych nie jest wymagane dostosowanie dawki (patrz punkt 5.2).
Zaburzenia czynności wątroby (profilaktyka udaru mózgu u pacjentów z migotaniem przedsionków, ZŻG/ZP)
Pacjenci z aktywnością enzymów wątrobowych dwukrotnie przekraczającą górną granicę normy (GGN) byli wykluczeni z kluczowych badań klinicznych. Nie ma doświadczenia w leczeniu tej subpopulacji pacjentów, dlatego nie zaleca się stosowania produktu Pradaxa w tej populacji (patrz punkty 4.4 i 5.2). Jest przeciwwskazany w przypadku zaburzeń czynności wątroby lub chorób wątroby, które mogą mieć wpływ na przeżycie (patrz punkt 4.3).
Przełączanie (profilaktyka udarów u pacjentów z migotaniem przedsionków, ZŻG/ZP)
Od leczenia lekiem Pradaxa do pozajelitowego leku przeciwzakrzepowego
Zaleca się odczekanie 12 godzin po podaniu ostatniej dawki przed zmianą eteksylanu dabigatranu na pozajelitowy lek przeciwzakrzepowy (patrz punkt 4.5).
Od pozajelitowych leków przeciwzakrzepowych po Pradaxa
Wstrzymać podawanie leków przeciwzakrzepowych do podawania pozajelitowego i rozpocząć podawanie eteksylanu dabigatranu na 0-2 godziny przed zaplanowaną kolejną dawką pierwotnego leczenia lub po przerwaniu leczenia w przypadku ciągłego leczenia (np. dożylna heparyna niefrakcjonowana (ENF)) (patrz punkt 4.5).
Od Pradaxy do antagonistów witaminy K (AVK)
Rozpoczęcie leczenia VKA należy ustalić na podstawie CRC według następujących wskazań:
• CrCL ≥ 50 ml/min, rozpocząć VKA 3 dni przed odstawieniem eteksylanu dabigatranu
• CrCL ≥ 30-
Ponieważ Pradaxa może zwiększyć wartość INR, będzie lepiej odzwierciedlać działanie AVK dopiero po upływie co najmniej 2 dni od zaprzestania stosowania leku Pradaxa.Do tego czasu wartości INR należy interpretować z ostrożnością.
Od AVK do Pradaxa
Należy przerwać stosowanie preparatu AVK Eteksylan dabigatranu można podawać, gdy tylko międzynarodowy współczynnik znormalizowany (INR)
Kardiowersja (profilaktyka udaru mózgu u pacjentów z migotaniem przedsionków, ZŻG/ZP)
Pacjenci poddawani kardiowersji mogą kontynuować leczenie dabigatranem.
Populacja pediatryczna (profilaktyka udaru mózgu u pacjentów z migotaniem przedsionków)
Stosowanie produktu Pradaxa u dzieci i młodzieży nie jest właściwe we wskazaniu: zapobieganie udarom mózgu i zatorowości systemowej u pacjentów z NVAF.
Populacja pediatryczna (ZŻG / ZP)
Bezpieczeństwo i skuteczność produktu Pradaxa u dzieci od urodzenia do 18 lat nie zostały jeszcze ustalone.Obecnie dostępne dane opisano w punktach 4.8 i 5.1, ale nie można sformułować zaleceń dotyczących dawkowania.
Pominięta dawka (profilaktyka udarów u pacjentów z migotaniem przedsionków, ZŻG/ZP)
Pominiętą dawkę eteksylanu dabigatranu można przyjąć do 6 godzin przed przyjęciem kolejnej dawki, po czym pominiętą dawkę należy pominąć.
Nie należy podwajać dawek w celu uzupełnienia pominiętej dawki.
Sposób podawania (prewencja pierwotna epizodów żylnej choroby zakrzepowo-zatorowej w chirurgii ortopedycznej, profilaktyka udaru mózgu u pacjentów z migotaniem przedsionków, ZŻG/ZP)
Lek Pradaxa można przyjmować z jedzeniem lub bez jedzenia. Pradaxa należy połykać w całości, popijając szklanką wody, aby ułatwić uwolnienie z żołądka.
Należy pouczyć pacjentów, aby nie otwierali kapsułek, ponieważ może to prowadzić do zwiększonego ryzyka krwawienia (patrz punkty 5.2 i 6.6).
04.3 Przeciwwskazania
• Nadwrażliwość na substancję czynną lub na którąkolwiek substancję pomocniczą wymienioną w punkcie 6.1
• Pacjenci z ciężkimi zaburzeniami czynności nerek (CrCL
• Klinicznie istotne aktywne krwawienie
• Urazy lub stany, jeśli są uważane za istotny czynnik ryzyka poważnego krwawienia. Mogą one obejmować obecne lub niedawno przebyte wrzody przewodu pokarmowego, wysokie ryzyko krwawień nowotworowych, niedawne uszkodzenie mózgu lub kręgosłupa, niedawno przebytą operację mózgu, kręgosłupa lub oka, niedawny krwotok śródczaszkowy, znane lub podejrzewane żylaki przełyku, malformacje tętniczo-żylne, tętniaki naczyń lub duże naczynia śródrdzeniowe lub śródmózgowe anomalie
• Jednoczesne leczenie innymi lekami przeciwzakrzepowymi, takimi jak niefrakcjonowana heparyna (ENF), heparyna drobnocząsteczkowa (enoksaparyna, dalteparyna itp.), pochodnymi heparyny (fondaparinuks itp.), doustnymi lekami przeciwzakrzepowymi (warfaryna, rywaroksaban, apiksaban itp.) okoliczności zmiany leczenia przeciwzakrzepowego (patrz punkt 4.2) lub kiedy ENF jest podawany w dawkach niezbędnych do utrzymania drożności cewnika do żyły centralnej lub tętnicy (patrz punkt 4.5)
• Zaburzenia czynności wątroby lub choroby wątroby, które mogą mieć jakikolwiek wpływ na przeżycie
• Jednoczesne leczenie układowym ketokonazolem, cyklosporyną, itrakonazolem i dronedaronem (patrz punkt 4.5)
• Protezy zastawki serca wymagające leczenia przeciwzakrzepowego (patrz punkt 5.1).
04.4 Specjalne ostrzeżenia i odpowiednie środki ostrożności dotyczące stosowania
Zaburzenia czynności wątroby
Pacjenci z podwyższoną aktywnością enzymów wątrobowych ponad dwukrotnie wyższą od górnej granicy normy byli wykluczeni z kluczowych badań klinicznych. Brak doświadczenia w leczeniu tej subpopulacji pacjentów i dlatego nie zaleca się stosowania produktu Pradaxa w tej populacji.
Ryzyko krwawienia
Eteksylan dabigatranu należy stosować ostrożnie w stanach zwiększonego ryzyka krwawienia oraz w sytuacjach związanych z jednoczesnym stosowaniem z substancjami zmieniającymi hemostazę poprzez hamowanie agregacji płytek krwi Krwawienie może wystąpić w dowolnym miejscu ciała podczas leczenia podczas stosowania eteksylanu dabigatranu Niewyjaśniony spadek stężenia hemoglobiny i / lub hematokryt lub ciśnienie krwi powinny skłonić do poszukiwania miejsca krwawienia.
Czynniki takie jak zmniejszona czynność nerek (30-50 ml/min CrCL), wiek ≥ 75 lat, mała masa ciała dabigatranu w osoczu (patrz punkty 4.2, 4.5 i 5.2).
Jednoczesne stosowanie tikagreloru zwiększa ekspozycję na dabigatran i może prowadzić do interakcji farmakodynamicznych, które mogą skutkować zwiększonym ryzykiem krwawienia (patrz punkt 4.5).
W badaniu dotyczącym zapobiegania udarom mózgu i SE u dorosłych pacjentów z NVAF eteksylan dabigatranu wiązał się z większą częstością występowania poważnych krwawień z przewodu pokarmowego, co było statystycznie istotne dla eteksylanu dabigatranu w dawce 150 mg dwa razy na dobę. Stosowanie kwasu acetylosalicylowego (ASA), klopidogrelu lub niesteroidowych leków przeciwzapalnych (NLPZ) oraz obecność zapalenia przełyku, żołądka lub refluksu żołądkowo-przełykowego również zwiększają ryzyko krwawienia z przewodu pokarmowego. U tych pacjentów z migotaniem przedsionków należy rozważyć dawkę 220 mg przyjmowaną jako jedna kapsułka 110 mg dwa razy na dobę i stosować dawkowanie zalecane w punkcie 4.2. Można rozważyć podanie PPI, aby zapobiec krwawieniu z przewodu pokarmowego.
Ryzyko krwawienia może być zwiększone u pacjentów jednocześnie leczonych selektywnymi inhibitorami wychwytu zwrotnego serotoniny (SSRI) lub selektywnymi inhibitorami wychwytu zwrotnego serotoniny i noradrenaliny (SNRI) (patrz punkt 4.5).
Podczas leczenia zaleca się ścisłą obserwację kliniczną (szukanie oznak krwawienia lub niedokrwistości), zwłaszcza w przypadku zbiegu czynników ryzyka (patrz punkt 5.1).
W tabeli 1 zestawiono czynniki, które mogą zwiększać ryzyko krwawienia. Patrz również przeciwwskazania w punkcie 4.3.
Tabela 1: Czynniki, które mogą zwiększać ryzyko krwawienia
Obecność zmian, stanów, zabiegów i (lub) leczenia lekami (takimi jak NLPZ, leki przeciwpłytkowe, SSRI i SNRI, patrz punkt 4.5), które znacząco zwiększają ryzyko poważnego krwawienia, wymaga starannej oceny stosunku korzyści do ryzyka. podawać tylko wtedy, gdy korzyści przewyższają ryzyko krwawienia.
Pradaxa zwykle nie wymaga rutynowego monitorowania parametrów krzepnięcia. Jednak ocena działania przeciwzakrzepowego związanego z dabigatranem może być przydatna w celu uniknięcia nadmiernie wysokiego narażenia na dabigatran w obecności dodatkowych czynników ryzyka. Test INR nie jest wiarygodny u pacjentów leczonych preparatem Pradaxa i zgłaszano fałszywie dodatnie zwiększenie INR, dlatego nie należy wykonywać testu INR Czas trombinowy w rozcieńczeniu osocza (dTT), czas ekarynowy (ECT), czas częściowej tromboplastyny po aktywacji (aPTT) może dostarczyć użytecznych informacji, ale testy nie są wystandaryzowane, a wyniki należy interpretować z ostrożnością (patrz punkt 5.1).
W Tabeli 2 przedstawiono wartości graniczne w minimalnym czasie testów krzepnięcia, które mogą wiązać się ze zwiększonym ryzykiem krwawienia (patrz punkt 5.1).
Tabela 2: Progowe wartości graniczne w minimalnym czasie testów krzepnięcia, które mogą wiązać się ze zwiększonym ryzykiem krwawienia
Pacjenci, u których wystąpi ostra niewydolność nerek, powinni przerwać przyjmowanie leku Pradaxa (patrz punkt 4.3).
Dane u pacjentów z wagą
W przypadku wystąpienia ciężkiego krwawienia leczenie należy przerwać i zbadać źródło krwawienia (patrz punkt 4.9).
Produkty lecznicze, które mogą zwiększać ryzyko krwotoku, nie powinny być podawane jednocześnie lub powinny być podawane z ostrożnością z produktem Pradaxa (patrz punkt 4.5).
Stosowanie leków fibrynolitycznych w leczeniu ostrego udaru niedokrwiennego mózgu
Można rozważyć zastosowanie fibrynolitycznych produktów leczniczych w leczeniu ostrego udaru niedokrwiennego mózgu, jeśli u pacjenta dTT, ECT lub APTT są poniżej górnej granicy normy, zgodnie z lokalnym zakresem referencyjnym.
Interakcje z induktorami P-gp
W przypadku równoczesnego podawania induktorów P-gp (takich jak ryfampicyna, ziele dziurawca zwyczajnego (Hypericum perforatum), karbamazepina lub fenytoina) można spodziewać się zmniejszenia stężenia dabigatranu w osoczu i dlatego należy tego unikać (patrz punkty 4.5 i 5.2).
Chirurgia i interwencje
Pacjenci przyjmujący eteksylan dabigatranu poddawani zabiegom chirurgicznym lub zabiegom inwazyjnym są narażeni na zwiększone ryzyko krwawienia. Dlatego interwencje chirurgiczne mogą wymagać czasowego zawieszenia leczenia.
Zaleca się zachowanie ostrożności i monitorowanie działania przeciwzakrzepowego w przypadku czasowego zawieszenia leczenia z powodu zabiegu chirurgicznego.Usunięcie dabigatranu u pacjentów z niewydolnością nerek może trwać dłużej (patrz punkt 5.2).Należy to ocenić przed każdym zabiegiem.W takich przypadkach należy wykonać test krzepnięcia (patrz punkty 4.4 i 5.1) mogą pomóc w ustaleniu, czy hemostaza jest nadal zaburzona.
Faza przedoperacyjna
Tabela 3 podsumowuje zasady wycofywania przed zabiegami inwazyjnymi lub chirurgicznymi.
Tabela 3: Zasady odstawienia przed zabiegami inwazyjnymi lub chirurgicznymi
Jeśli konieczne jest pilne działanie, eteksylan dabigatranu należy tymczasowo zawiesić. W miarę możliwości operację/interwencję należy odroczyć co najmniej do 12 godzin po przyjęciu ostatniej dawki. Jeśli nie można odroczyć zabiegu chirurgicznego, może wystąpić zwiększone ryzyko krwawienia.Ryzyko krwawienia należy rozważyć w stosunku do pilności zabiegu chirurgicznego (kardiowersja, patrz punkt 4.2).
Znieczulenie podpajęczynówkowe / znieczulenie zewnątrzoponowe / nakłucie lędźwiowe
Procedury takie jak znieczulenie podpajęczynówkowe wymagają normalnych funkcji hemostatycznych.
Ryzyko wystąpienia krwiaka rdzeniowego lub nadtwardówkowego może być zwiększone w przypadku urazowego lub wielokrotnego nakłucia oraz przy długotrwałym stosowaniu cewników zewnątrzoponowych.Po usunięciu cewnika, przed podaniem pierwszej dawki eteksylanu dabigatranu powinien upłynąć co najmniej 2-godzinny odstęp. Pacjenci ci wymagają częstej obserwacji objawów neurologicznych krwiaka rdzeniowego lub nadtwardówkowego.
Faza pooperacyjna
Podawanie eteksylanu dabigatranu należy wznowić tak szybko, jak to możliwe po zabiegu inwazyjnym lub zabiegu chirurgicznym, pod warunkiem, że ustalono, że sytuacja kliniczna pozwala na odpowiednią hemostazę.
Pacjenci z wysokim ryzykiem krwawienia lub pacjenci z ryzykiem nadmiernej ekspozycji, szczególnie pacjenci z umiarkowanymi zaburzeniami czynności nerek (CrCL 30-50 ml/min), powinni być leczeni z ostrożnością (patrz punkty 4.4 i 5.1).
Pacjenci z wysokim ryzykiem zgonu z powodu operacji i z wewnętrznymi czynnikami ryzyka zdarzeń zakrzepowo-zatorowych
Dane dotyczące skuteczności i bezpieczeństwa stosowania dabigatranu u tych pacjentów są ograniczone i dlatego należy zachować ostrożność podczas leczenia.
Operacja złamania biodra
Brak danych dotyczących stosowania produktu Pradaxa u pacjentów poddawanych operacji złamania szyjki kości udowej. Dlatego leczenie nie jest zalecane.
Zawał mięśnia sercowego (profilaktyka udaru mózgu u pacjentów z migotaniem przedsionków)
W badaniu III fazy RE-LY (patrz punkt 5.1) całkowita częstość występowania zawału mięśnia sercowego (MI) wynosiła odpowiednio 0,82, 0,81 i 0,64%/rok odpowiednio dla eteksylanu dabigatranu 110 mg dwa razy na dobę, eteksylanu dabigatranu 150 mg dwa razy na dobę i warfaryny, z względne zwiększenie ryzyka dla dabigatranu o 29% i 27% w porównaniu z warfaryną Niezależnie od zastosowanej terapii, największe bezwzględne ryzyko MI obserwowano w następujących podgrupach, przy podobnym ryzyku względnym: pacjenci z wcześniejszym MI, pacjenci ≥ 65 lat wiek z cukrzycą lub chorobą wieńcową, pacjenci z frakcją wyrzutową lewej komory
Zawał mięśnia sercowego (ZŻG/ZP)
W trzech aktywnie kontrolowanych badaniach klinicznych zgłoszono większą częstość występowania MI u pacjentów leczonych eteksylanem dabigatranu niż u pacjentów leczonych warfaryną: 0,4% w porównaniu z 0,2% w krótkoterminowych badaniach RE-COVER i RE-COVER II; oraz 0,8% w porównaniu z 0,1% w długoterminowym badaniu RE-MEDY. W tym badaniu wzrost był istotny statystycznie (p = 0,022).
W badaniu RE-SONATE, w którym porównywano eteksylan dabigatranu z placebo, częstość występowania MI wyniosła 0,1% u pacjentów przyjmujących eteksylan dabigatranu i 0,2% u pacjentów otrzymujących placebo.
Pacjenci z czynną chorobą nowotworową (ZŻG/ZP)
Skuteczność i bezpieczeństwo nie zostały ustalone dla DVT / PE u pacjentów z istniejącym nowotworem.
Barwniki
Kapsułki twarde Pradaxa zawierają żółcień pomarańczową (E110), która może powodować reakcje alergiczne.
04.5 Interakcje z innymi produktami leczniczymi i inne formy interakcji
Antykoagulanty i leki przeciwpłytkowe
Brak lub ograniczone doświadczenie dotyczące następujących metod leczenia, które mogą zwiększać ryzyko krwawienia w przypadku jednoczesnego stosowania z produktem Pradaxa: leki przeciwzakrzepowe, takie jak heparyna niefrakcjonowana (ENF), heparyna drobnocząsteczkowa (LMWH) i pochodne heparyny (fondaparinux, desirudyna), leki trombolityczne produkty lecznicze i antagoniści witaminy K, rywaroksaban lub inne doustne leki przeciwzakrzepowe (patrz punkt 4.3) oraz leki przeciwpłytkowe, takie jak antagoniści receptora GPIIb/IIIa, tiklopidyna, prasugrel, tikagrelor, dekstran i sulfinpirazon (patrz punkt 4.4).
Na podstawie ograniczonych danych zebranych w badaniu III fazy RE-LY u pacjentów z migotaniem przedsionków zaobserwowano, że jednoczesne stosowanie innych doustnych lub pozajelitowych leków przeciwzakrzepowych zwiększało około 2,5-krotnie częstość występowania poważnych krwawień, zarówno w przypadku eteksylanu dabigatranu, niż w przypadku warfaryny. , zwłaszcza w przypadku zmiany jednego leku przeciwzakrzepowego na inny (patrz punkt 4.3).
ENF można podawać w dawkach niezbędnych do utrzymania drożności cewnika do żyły centralnej lub tętnicy (patrz punkt 4.3).
Klopidogrel i ASANa podstawie danych zebranych w badaniu III fazy RE-LY (patrz punkt 5.1) zaobserwowano, że jednoczesne stosowanie leków przeciwpłytkowych, ASA lub klopidogrelu, w przybliżeniu podwaja częstość występowania poważnych krwawień zarówno w przypadku eteksylanu dabigatranu, jak i warfaryny (patrz punkt 4.4).
Klopidogrel: W badaniu I fazy z udziałem zdrowych, młodych ochotników płci męskiej jednoczesne podawanie eteksylanu dabigatranu i klopidogrelu nie powodowało dalszego wydłużenia czasu krwawienia z naczyń włosowatych w porównaniu z samym klopidogrelem. Ponadto „AUCa, Ss i Cmax, ss i miary krzepnięcia dla wpływu dabigatranu lub hamowania agregacji płytek krwi jako miary działania klopidogrelu” pozostały zasadniczo niezmienione przy porównywaniu leczenia skojarzonego i odpowiedniego dawka nasycająca 300 mg lub 600 mg klopidogrelu, AUC, ss i Cmax, ss dabigatranu zwiększyły się o około 30-40% (patrz punkt 4.4) (patrz także podpunkt poniżej dotyczący „ASA”).
ASA: Wpływ równoczesnego podawania eteksylanu dabigatranu i ASA na ryzyko krwawienia badano u pacjentów z migotaniem przedsionków w badaniu II fazy, w którym zastosowano randomizowane jednoczesne podawanie ASA. jednoczesne podawanie ASA i eteksylanu dabigatranu w dawce 150 mg dwa razy na dobę może zwiększać ryzyko dowolnego rodzaju krwawienia z 12% do 18% i 24% przy odpowiednio 81 mg i 325 mg ASA (patrz punkt 4.4).
NLPZ: Wykazano, że NLPZ podawane jako krótko działające leki przeciwbólowe w okresie okołooperacyjnym nie są związane ze zwiększonym ryzykiem krwawienia w połączeniu z eteksylanem dabigatranu.Przewlekłe stosowanie NLPZ zwiększa ryzyko krwawienia o około 50%. i warfaryna. Dlatego, ze względu na ryzyko krwotoku, szczególnie w przypadku NLPZ o okresie półtrwania w fazie eliminacji > 12 godzin, zaleca się ścisłą obserwację objawów krwawienia (patrz punkt 4.4).
HDCz: Jednoczesne stosowanie HDCz, takich jak enoksaparyna i eteksylan dabigatranu, nie zostało konkretnie ocenione.Po zmianie leczenia z 3-dniowego leczenia enoksaparyną w dawce 40 mg raz dziennie drogą sc, 24 godziny po podaniu HDCz. enoksaparyny, ekspozycja na dabigatran była nieco mniejsza niż po podaniu samego eteksylanu dabigatranu (pojedyncza dawka 220 mg). Większą aktywność anty-FXa / FIIa obserwowano po podaniu eteksylanu dabigatranu poprzedzonego wstępnym leczeniem enoksaparyną w porównaniu z leczeniem dabigatranem Uważa się, że jest to spowodowane napędzającym działaniem leczenia enoksaparyną i uważa się, że nie ma to znaczenia klinicznego.Wyniki innych testów działania przeciwzakrzepowego związanego z dabigatranem nie uległy istotnej zmianie w wyniku wstępnego leczenia enoksaparyną.
Interakcje związane z profilem metabolicznym eteksylanu dabigatranu i dabigatranu
Eteksylan dabigatranu i dabigatran nie są metabolizowane przez układ cytochromu P450 i nie wywołują żadnego efektu. in vitro na ludzkie enzymy cytochromu P450. Dlatego nie oczekuje się interakcji z powiązanymi produktami leczniczymi i dabigatranem.
Interakcje transportera
Inhibitory P-gp
Eteksylan dabigatranu jest substratem transportera wypływowego P-gp. Jednoczesne podawanie z inhibitorami P-gp (takimi jak amiodaron, werapamil, chinidyna, ketokonazol, dronedaron, klarytromycyna i tikagrelor) może powodować zwiększenie stężenia dabigatranu w osoczu.
O ile nie zalecono inaczej, w przypadku jednoczesnego podawania dabigatranu z silnymi inhibitorami P-gp wymagane jest ścisłe monitorowanie kliniczne (obserwacja objawów krwawienia lub niedokrwistości). Test krzepnięcia pomaga zidentyfikować pacjentów ze zwiększonym ryzykiem krwawienia z powodu „zwiększonej ekspozycji na dabigatran (patrz punkty 4.2, 4.4 i 5.1).
Następujące silne inhibitory P-gp są przeciwwskazane: ogólnoustrojowo podawany ketokonazol, cyklosporyna, itrakonazol i dronedaron (patrz punkt 4.3) Nie zaleca się jednoczesnego leczenia takrolimusem Inhibitory P-gp pochodzące ze słabych leków o umiarkowanym nasileniu (np. amiodaron, pozakonazol, chinidyna, werapamil) i tikagrelor) należy stosować ostrożnie (patrz punkty 4.2 i 4.4).
Ketokonazol: Ketokonazol po podaniu pojedynczej dawki doustnej 400 mg zwiększał całkowite AUC0-∞ i Cmax dabigatranu odpowiednio o 138% i 135% oraz o 153% i 149% po wielokrotnych doustnych dawkach 400 mg ketokonazolu jednorazowo codzienny. Ketokonazol nie zmieniał czasu do osiągnięcia maksimum, końcowego okresu półtrwania i średniego czasu przebywania (patrz punkt 4.4) Jednoczesne stosowanie z ogólnoustrojowym ketokonazolem jest przeciwwskazane (patrz punkt 4.3).
Dronedaron: Podczas jednoczesnego podawania eteksylanu dabigatranu i dronedaronu całkowite wartości AUC0-∞ i Cmax dabigatranu wzrosły odpowiednio około 2,4-krotnie i 2,3-krotnie (+136% i 125%) po wielokrotnych dawkach 400. mg dronedaronu dwa razy na dobę i odpowiednio około 2,1-krotność i 1,9-krotność (+114% i 87%), po podaniu pojedynczej dawki 400 mg Na końcowy okres półtrwania i klirens nerkowy dabigatranu dronedaron nie miał wpływu. dawki dronedaronu podawano 2 godziny po podaniu eteksylanu dabigatranu, zwiększenie AUC0-∞ dabigatranu było odpowiednio 1,3-krotne i 1,6-krotne. Jednoczesne leczenie dronedaronem jest przeciwwskazane.
Amiodaron: W przypadku jednoczesnego podawania produktu Pradaxa z pojedynczą doustną dawką 600 mg amiodaronu ilość i szybkość wchłaniania amiodaronu i jego aktywnego metabolitu DEA zasadniczo nie uległy zmianie. AUC i Cmax dabigatranu wzrosły odpowiednio o około 60% i 50% Mechanizm interakcji nie jest w pełni wyjaśniony. Biorąc pod uwagę długi okres półtrwania amiodaronu, potencjalne interakcje mogą utrzymywać się przez kilka tygodni po odstawieniu amiodaronu (patrz punkty 4.2 i 4.4).
U pacjentów leczonych w celu zapobiegania żylnej chorobie zakrzepowo-zatorowej po operacji wymiany stawu biodrowego lub kolanowego, dawkę produktu Pradaxa należy zmniejszyć do 150 mg raz na dobę w postaci 2 kapsułek po 75 mg w przypadku jednoczesnego leczenia eteksylanem dabigatranu i amiodaronem (patrz punkt 4.2). Zaleca się ścisłą obserwację kliniczną, gdy eteksylan dabigatranu jest podawany w skojarzeniu z amiodaronem, szczególnie w przypadku wystąpienia krwawienia oraz z zachowaniem szczególnej ostrożności w przypadku pacjentów z łagodnymi lub umiarkowanymi zaburzeniami czynności nerek.
Chinidyna: Chinidynę podawano w dawkach 200 mg co 2 godziny do łącznej dawki 1000 mg. Eteksylan dabigatranu podawano dwa razy dziennie przez 3 kolejne dni, trzeciego dnia z chinidyną lub bez. AUC, ss i Cmax, ss dabigatranu zwiększały się średnio odpowiednio o 53% i 56% podczas jednoczesnego podawania chinidyny (patrz punkty 4.2 i 4.4).
U pacjentów leczonych w celu zapobiegania żylnej chorobie zakrzepowo-zatorowej po operacji wymiany stawu biodrowego lub kolanowego, dawkę produktu Pradaxa należy zmniejszyć do 150 mg raz na dobę w postaci 2 kapsułek po 75 mg, jeśli są leczeni jednocześnie z eteksylanem dabigatranu i chinidyną (patrz punkt 4.2). Zaleca się ścisłą obserwację kliniczną, gdy eteksylan dabigatranu jest podawany w skojarzeniu z chinidyną, szczególnie w przypadku wystąpienia krwawienia oraz z zachowaniem szczególnej ostrożności w przypadku pacjentów z łagodnymi lub umiarkowanymi zaburzeniami czynności nerek.
Werapamil: Gdy eteksylan dabigatranu (150 mg) był podawany jednocześnie z werapamilem doustnym, Cmax i AUC dabigatranu wzrastały, ale wielkość tej zmiany różniła się w zależności od czasu podania i postaci werapamilu (patrz punkty 4.2 i 4.4).
Maksymalny wzrost ekspozycji na dabigatran zaobserwowano po podaniu pierwszej dawki werapamilu w postaci o natychmiastowym uwalnianiu, podanej godzinę przed przyjęciem eteksylanu dabigatranu (wzrost Cmax o około 180% i AUC o około 150%). Działanie stopniowo zmniejszało się po podaniu postaci o przedłużonym uwalnianiu (wzrost Cmax o około 90% i AUC o około 70%) lub po podaniu wielokrotnych dawek werapamilu (wzrost Cmax o około 60% i zwiększenie AUC o około 50%).
Dlatego w przypadku jednoczesnego podawania dabigatranu z werapamilem konieczne jest uważne monitorowanie kliniczne (poszukiwanie oznak krwawienia lub niedokrwistości). U pacjentów z prawidłową czynnością nerek po operacji wymiany stawu biodrowego lub kolanowego leczonych jednocześnie eteksylanem dabigatranu i werapamilem, dawkę produktu Pradaxa należy zmniejszyć do 150 mg przyjmowanych w postaci 2 kapsułek po 75 mg raz na dobę. eteksylanu dabigatranu i werapamilu, należy rozważyć zmniejszenie dawki produktu Pradaxa do 75 mg na dobę (patrz punkty 4.2 i 4.4).
W przypadku pacjentów z NLPZ leczonych w celu zapobiegania udarowi i SE oraz pacjentów z ZŻG/ZP leczonych eteksylanem dabigatranu i werapamilem, dawkę produktu Pradaxa należy zmniejszyć do 220 mg przyjmowanych jako jedna kapsułka 110 mg dwa razy na dobę (patrz punkt 4.2).
Zaleca się ścisłą obserwację kliniczną, gdy eteksylan dabigatranu jest skojarzony z werapamilem, szczególnie w przypadku wystąpienia krwawienia oraz z zachowaniem szczególnej ostrożności w przypadku pacjentów z łagodnymi lub umiarkowanymi zaburzeniami czynności nerek.
Nie zaobserwowano znaczących interakcji po podaniu werapamilu 2 godziny po przyjęciu eteksylanu dabigatranu (wzrost Cmax o około 10% i wzrost AUC o około 20%), co tłumaczy się całkowitym wchłanianiem dabigatranu po 2 godzinach (patrz punkt 4.4).
Klarytromycyna: Gdy klarytromycyna (500 mg dwa razy na dobę) była podawana zdrowym ochotnikom w skojarzeniu z eteksylanem dabigatranu, obserwowano zwiększenie AUC o około 19% i Cmax o około 15% bez wpływu na bezpieczeństwo kliniczne. dabigatran, nie można wykluczyć klinicznie istotnej interakcji w przypadku skojarzenia z klarytromycyną. Dlatego należy uważnie monitorować, gdy eteksylan dabigatranu jest skojarzony z klarytromycyną, a zwłaszcza w przypadku krwawienia, zwłaszcza u pacjentów z łagodnymi lub umiarkowanymi zaburzeniami czynności nerek.
Tikagrelor: Gdy pojedynczą dawkę 75 mg eteksylanu dabigatranu podano jednocześnie z dawką początkową 180 mg tikagreloru, AUC i Cmax dabigatranu wzrosły odpowiednio o 1,73 i 1,95-krotnie (+73 % i 95%). mg tikagreloru dwa razy dziennie zwiększenie ekspozycji na dabigatran jest 1,56 i 1,46-krotne (+56% i 46%) odpowiednio dla AUC i Cmax.
Jednoczesne podanie początkowej dawki 180 mg tikagreloru i 110 mg eteksylanu dabigatranu (w stanie stacjonarnym) zwiększyło AUCa, Ss i Cmax, ss dabigatranu odpowiednio 1,49-krotnie i 1,65-krotnie (+ 49% i 65 %), w porównaniu z podawaniem samego eteksylanu dabigatranu. Gdy początkową dawkę 180 mg tikagreloru podano 2 godziny po podaniu 110 mg eteksylanu dabigatranu (stan stacjonarny), wzrost AUCa, Ss i Cmax, ss dabigatranu zmniejszył się odpowiednio 1,27-krotnie i 1,23 razy (+ 27% i 23% w porównaniu z podawaniem samego eteksylanu dabigatranu. To rozłożone w czasie podawanie jest zalecane do rozpoczynania leczenia tikagrelorem od dawki początkowej.
Jednoczesne podawanie 90 mg tikagreloru dwa razy na dobę (dawka podtrzymująca) z 110 mg eteksylanu dabigatranu zwiększało skorygowane wartości AUCa, Ss i Cmax, ss dabigatranu odpowiednio 1,26-krotnie i 1,29-krotnie w porównaniu z podaniem samego eteksylanu dabigatranu.
Następujące silne inhibitory P-gp nie były badane klinicznie, ale na podstawie danych: in vitro oczekuje się działania podobnego do działania ketokonazolu:
Itrakonazol i cyklosporyna, które są przeciwwskazane (patrz punkt 4.3).
Wykazano, że takrolimus in vitro ma podobny wpływ hamujący na P-gp jak itrakonazol i cyklosporyna. Eteksylan dabigatranu nie był badany klinicznie w skojarzeniu z takrolimusem. Jednak ograniczone dane kliniczne dostępne dla innego substratu P-gp (ewerolimusu) sugerują, że hamowanie P-gp przez takrolimus jest słabsze niż obserwowane w przypadku silnych inhibitorów P-gp. Na podstawie tych danych nie zaleca się jednoczesnego leczenia takrolimusem .
Pozakonazol również częściowo hamuje P-gp, ale nie był badany klinicznie. Należy zachować ostrożność podczas jednoczesnego podawania produktu Pradaxa i pozakonazolu.
Induktory P-gp
Jednoczesne podawanie induktora P-gp (takiego jak ryfampicyna, ziele dziurawca zwyczajnego (Hypericum perforatum), karbamazepina lub fenytoina) może zmniejszać stężenie dabigatranu i należy tego unikać (patrz punkty 4.4 i 5.2).
Ryfampicyna: Wstępne podanie induktora ryfampicyny w dawce 600 mg raz na dobę przez 7 dni zmniejszyło całkowity szczyt dabigatranu i całkowitą ekspozycję odpowiednio o 65,5% i 67%. Efekt induktora uległ zmniejszeniu, powodując ekspozycję na dabigatran zbliżoną do wartości referencyjnej w ciągu siódmego dnia po zaprzestaniu leczenia ryfampicyną. Po kolejnych 7 dniach nie zaobserwowano zwiększenia biodostępności.
Inne leki wpływające na P-gp
Inhibitory proteazy, takie jak rytonawir i jego kombinacje z innymi inhibitorami proteazy, wpływają na P-gp (zarówno jako inhibitory, jak i induktory). Ponieważ nie zostały zbadane, nie zaleca się ich jednoczesnego leczenia z produktem Pradaxa.
Substrat P-gp
Digoksyna: W badaniu z udziałem 24 zdrowych pacjentów, w którym produkt Pradaxa podawano w skojarzeniu z digoksyną, nie zaobserwowano ani zmian w digoksynie, ani istotnych zmian klinicznych w ekspozycji na dabigatran.
Jednoczesne stosowanie selektywnych inhibitorów wychwytu zwrotnego serotoniny (SSRI) lub selektywnych inhibitorów wychwytu zwrotnego serotoniny i noradrenaliny (SNRI)
SSRI i SNRI zwiększały ryzyko krwawienia we wszystkich leczonych grupach w badaniu RE-LY.
pH żołądka
Pantoprazol: Gdy lek Pradaxa podawano w skojarzeniu z pantoprazolem, obserwowano około 30% zmniejszenie powierzchni pod krzywą stężenia dabigatranu w osoczu w funkcji czasu.Obserwowano pantoprazol i inne inhibitory pompy protonowej (PPI). jednoczesne leczenie PPI nie wydawało się zmniejszać skuteczności produktu Pradaxa.
Ranitydyna: Podawanie ranitydyny z produktem Pradaxa nie ma klinicznie istotnego wpływu na wchłanianie dabigatranu.
04.6 Ciąża i laktacja
Kobiety w wieku rozrodczym / Antykoncepcja dla mężczyzn i kobiet
Kobiety w wieku rozrodczym powinny unikać zajścia w ciążę podczas leczenia eteksylanem dabigatranu.
Ciąża
Dostępna jest ograniczona ilość danych dotyczących stosowania eteksylanu dabigatranu u kobiet w ciąży.
Badania na zwierzętach wykazały szkodliwy wpływ na reprodukcję (patrz punkt 5.3). Potencjalne ryzyko dla ludzi nie jest znane.
Leku Pradaxa nie należy stosować w okresie ciąży, chyba że jest to bezwzględnie konieczne.
Czas karmienia
Brak danych klinicznych dotyczących wpływu dabigatranu na karmione niemowlęta.
Karmienie piersią należy przerwać na czas leczenia produktem Pradaxa.
Płodność
Brak danych u ludzi.
W badaniach na zwierzętach po dawce 70 mg/kg obserwowano wpływ na płodność samic w postaci zmniejszonej implantacji i zwiększonej utraty przedimplantacyjnej (ekspozycja w osoczu 5-krotnie większa niż u pacjentów). Nie zaobserwowano żadnego innego wpływu na płodność samic. Nie stwierdzono wpływu na płodność mężczyzn. Przy dawkach toksycznych dla matek (ekspozycja w osoczu od 5 do 10 razy większa niż u pacjentek) u szczurów i królików obserwowano zmniejszenie masy ciała płodu i żywotności zarodka i płodu z nasileniem zmian u płodu. W badaniach przed- i pourodzeniowych obserwowano zwiększoną śmiertelność płodów przy dawkach toksycznych dla matek (dawka odpowiadająca 4-krotnie większej ekspozycji w osoczu niż u pacjentów).
04.7 Wpływ na zdolność prowadzenia pojazdów i obsługiwania maszyn
Pradaxa nie ma wpływu lub ma nieistotny wpływ na zdolność prowadzenia pojazdów i obsługiwania maszyn.
04.8 Działania niepożądane
Podsumowanie profilu bezpieczeństwa
W 6 aktywnie kontrolowanych badaniach profilaktyki ŻChZZ leczono łącznie 10 795 pacjentów z użyciem co najmniej jednej mocy badanego leku. Spośród tych 6684 pacjentów leczono 150 mg lub 220 mg leku Pradaxa na dobę.
W głównym badaniu dotyczącym zapobiegania udarom mózgu i SE u pacjentów z migotaniem przedsionków eteksylanem dabigatranu leczono łącznie 12042 pacjentów, z czego 6059 otrzymywało 150 mg eteksylanu dabigatranu dwa razy na dobę, a 5983 było leczonych eteksylanem dabigatranu. otrzymali dawki 110 mg dwa razy na dobę.
W dwóch aktywnie kontrolowanych badaniach dotyczących leczenia ZŻG/ZP, RE-COVER i RE-COVER II, do analizy bezpieczeństwa eteksylanu dabigatranu włączono łącznie 2553 pacjentów.Wszyscy pacjenci otrzymywali eteksylan dabigatranu w dawce 150 mg dwa razy na dobę. w przypadku obu terapii eteksylan dabigatranu i warfarynę liczy się od pierwszego przyjęcia eteksylanu dabigatranu lub warfaryny po zaprzestaniu leczenia pozajelitowego (tylko w okresie leczenia doustnego) Obejmuje to wszystkie działania niepożądane, które wystąpiły podczas leczenia dabigatranem Wszystkie działania niepożądane, które wystąpiły podczas leczenia leczenie warfaryną jest włączone, z wyjątkiem tych, które wystąpiły podczas nakładania się warfaryny i leczenia pozajelitowego.
Łącznie 2114 pacjentów było leczonych w aktywnie kontrolowanym badaniu dotyczącym zapobiegania ZŻG/ZP RE-MEDY oraz badaniu dotyczącym zapobiegania ZŻG/ZP RE-SONATE. Wszyscy pacjenci otrzymywali eteksylan dabigatranu w dawce 150 mg dwa razy na dobę.
Łącznie około 9% pacjentów leczonych z powodu planowej operacji biodra lub kolana (leczenie krótkoterminowe do 42 dni), 22% pacjentów z migotaniem przedsionków leczonych z powodu udaru mózgu i SE (leczenie długoterminowe do 3 lat), 14 Działania niepożądane wystąpiły u % pacjentów leczonych z powodu ZŻG/ZP i 15% pacjentów leczonych w celu zapobiegania ZŻG/ZP.
Najczęściej zgłaszanymi działaniami niepożądanymi są krwawienia, które wystąpiły łącznie u około 14% pacjentów leczonych krótkoterminowo z powodu planowej alloplastyki stawu biodrowego lub kolanowego, u 16,6% pacjentów z długotrwałym migotaniem przedsionków. 14,4% pacjentów leczonych z powodu ZŻG/ZP Ponadto krwawienie wystąpiło u 19,4% pacjentów w badaniu profilaktyki ZŻG/ZP RE-MEDY i 10,5% pacjentów w badaniu profilaktyki ZŻG/ZP RE-SONATE.
Ponieważ populacje pacjentów leczonych w trzech wskazaniach nie są porównywalne, a zdarzenia krwotoczne są rozłożone na różne klasy układów narządowych (SOC), skrócony opis epizodów poważnych krwawień i wszelkiego rodzaju krwawień z podziałem na wskazania.
Chociaż zdarzało się to rzadko w badaniach klinicznych, mogą wystąpić poważne lub poważne krwawienia, które niezależnie od lokalizacji mogą prowadzić do kalectwa, zagrażać życiu, a nawet śmierci.
Tabela podsumowująca działania niepożądane
W tabeli 4 przedstawiono działania niepożądane zidentyfikowane w badaniach profilaktyki pierwotnej ŻChZZ po operacji wymiany stawu biodrowego lub kolanowego, w badaniu dotyczącym zapobiegania udarowi zakrzepowo-zatorowemu i ES u pacjentów z migotaniem przedsionków oraz w badaniach dotyczących leczenia i profilaktyki ZŻG/ZP zleconych przez SOC. i częstość, stosując następującą konwencję: bardzo często (≥ 1/10); często (≥ 1/100,
Tabela 4: Działania niepożądane
Prewencja pierwotna żylnych epizodów zakrzepowo-zatorowych w chirurgii ortopedycznej
Krwawienie
W tabeli 5 przedstawiono liczbę (%) pacjentów, u których wystąpiły niepożądane krwawienia w okresie leczenia profilaktycznego VTE w dwóch głównych badaniach klinicznych, w zależności od dawki.
Tabela 5: Liczba (%) pacjentów, u których wystąpiły niepożądane reakcje krwotoczne
Definicje działań niepożądanych dużego krwawienia w badaniach RE-NOVATE i RE-MODEL były następujące:
• śmiertelne krwawienie
• klinicznie manifestujące się krwawienie związane ze spadkiem stężenia hemoglobiny ≥ 20 g/l (co odpowiada 1,24 mmol/l), oba powyżej oczekiwanego
• klinicznie manifestują się krwawienie przekraczające oczekiwane i wymagające transfuzji ≥ 2 jednostek erytrocytów lub krwi pełnej powyżej oczekiwanej
• objawowe krwawienie zaotrzewnowe, śródczaszkowe, śródgałkowe lub śródrdzeniowe
• krwawienie wymagające przerwania leczenia
• krwawienie, które wymagało nowej operacji.
W przypadku krwawienia zaotrzewnowego (USG lub tomografia komputerowa (CT)) i krwawienia śródrdzeniowego (TK lub rezonans magnetyczny) wymagane były obiektywne testy.
Zapobieganie udarom i ES u dorosłych pacjentów z NLPZ z jednym lub więcej czynnikami ryzyka
Krwawienie
Tabela 6 przedstawia główne lub dowolne zdarzenia krwawienia stwierdzone w głównym badaniu oceniającym zapobieganie udarom mózgu i SE u pacjentów z migotaniem przedsionków.
Tabela 6: Przypadki krwawień w badaniu oceniającym zapobieganie udarom i SE u pacjentów z migotaniem przedsionków
Poważne krwawienie definiowano, spełniając co najmniej jedno z następujących kryteriów:
Krwawienie związane ze spadkiem stężenia hemoglobiny o co najmniej 20 g/l lub wymagające przetoczenia co najmniej 2 jednostek krwinek czerwonych lub krwi pełnej.
Krwawienie objawowe w krytycznym „obszarze lub narządzie”: wewnątrzgałkowe, wewnątrzczaszkowe, dordzeniowe lub domięśniowe z zespołem ciasnoty, krwawienie zaotrzewnowe, krwawienie śródstawowe lub krwawienie z osierdzia.
Poważne krwawienie klasyfikowano jako zagrażające życiu, jeśli spełniało co najmniej jedno z następujących kryteriów:
Krwawienie śmiertelne, objawowe krwawienie śródczaszkowe; zmniejszenie stężenia hemoglobiny o co najmniej 50 g/l przetoczenie co najmniej 4 jednostek erytrocytów lub pełnej krwi krwawienie związane z niedociśnieniem wymagające zastosowania leków inotropowych podawanych dożylnie krwawienie wymagające operacji.
Pacjenci przydzieleni losowo do grupy otrzymującej eteksylan dabigatranu 110 mg dwa razy na dobę lub 150 mg dwa razy na dobę mieli znacznie mniejsze ryzyko krwawienia zagrażającego życiu i krwawienia śródczaszkowego niż pacjenci leczeni warfaryną [p
Korzyści kliniczne ze stosowania eteksylanu dabigatranu w zakresie zapobiegania udarowi i SE oraz zmniejszenia ryzyka ICH w porównaniu z warfaryną utrzymują się w poszczególnych podgrupach, np. zaburzenia czynności nerek, wiek, jednoczesne stosowanie produktów leczniczych, takich jak leki przeciwagregacyjne lub inhibitory P-gp. niektóre podgrupy pacjentów są narażone na zwiększone ryzyko poważnego krwawienia podczas leczenia antykoagulantem, ryzyko nadmiernego krwawienia w przypadku eteksylanu dabigatranu jest spowodowane krwawieniem z przewodu pokarmowego, zwykle występującym w ciągu pierwszych 3-6 miesięcy po rozpoczęciu leczenia.
Leczenie zakrzepicy żył głębokich (ZŻG) i zatorowości płucnej (ZP) oraz zapobieganie nawrotom ZŻG i ZP u dorosłych (leczenie ZŻG/ZP).
Tabela 7 przedstawia zdarzenia krwawienia w połączonej analizie głównych badań RE-COVER i RE-COVER II, w których oceniano leczenie zakrzepicy żył głębokich (ZŻG) i zatorowości płucnej (ZP). krwawienia i jakiekolwiek krwawienia były znacznie niższe niż w przypadku warfaryny przy nominalnym poziomie alfa 5%.
Tabela 7: Przypadki krwawień w badaniach RE-COVER i RE-COVER II, w których oceniano leczenie zakrzepicy żył głębokich (ZŻG) i zatorowości płucnej (ZP)
Przypadki krwawienia w przypadku obu terapii są liczone od pierwszego przyjęcia eteksylanu dabigatranu lub warfaryny po przerwaniu leczenia pozajelitowego (tylko okres leczenia doustnego). Obejmuje to wszystkie krwawienia, które wystąpiły podczas leczenia eteksylanem dabigatranu. Uwzględniono wszystkie krwawienia, które wystąpiły podczas leczenia warfaryną, z wyjątkiem tych, które wystąpiły podczas nakładania się warfaryny i leczenia pozajelitowego.
Definicja poważnych krwawień była zgodna z zaleceniami Międzynarodowego Towarzystwa Zakrzepicy i Hemostazy. Krwawienie uznawano za poważne, jeśli spełniało co najmniej jedno z następujących kryteriów:
• Śmiertelne krwawienie
• Objawowe krwawienie w „krytycznym obszarze lub narządzie, takim jak wewnątrzczaszkowe, wewnątrzrdzeniowe, wewnątrzgałkowe, zaotrzewnowe, dostawowe, osierdziowe lub domięśniowe z zespołem ciasnoty kompartmentowej. z sytuacją. przychodnia objawowa
• Krwawienie, które spowodowało spadek poziomu hemoglobiny o 20 g/l (1,24 mmol/l) lub więcej lub które spowodowało przetoczenie 2 lub więcej jednostek krwi pełnej lub krwinek czerwonych.
W tabeli 8 przedstawiono zdarzenia krwawienia w głównym badaniu RE-MEDY, w którym oceniano profilaktykę zakrzepicy żył głębokich (ZŻG) i zatorowości płucnej (ZP).Niektóre krwawienia (poważne, klinicznie istotne i jakiekolwiek krwawienia) były znacznie mniejsze niż nominalna wartość alfa 5% u pacjentów leczonych eteksylanem dabigatranu w porównaniu z pacjentami leczonymi warfaryną.
Tabela 8: Przypadki krwawień w badaniu RE-MEDY, w którym oceniano profilaktykę zakrzepicy żył głębokich (ZŻG) i zatorowości płucnej (ZP)
* HR nie podlega ocenie, ponieważ nie ma zdarzenia w jednej z dwóch kohort/leczeń
Definicja poważnych krwawień była zgodna z zaleceniami Międzynarodowego Towarzystwa Zakrzepicy i Hemostazy, jak opisano dla RECOVER i RECOVER II.
W tabeli 9 przedstawiono zdarzenia krwawienia w głównym badaniu RE-SONATE, w którym oceniano profilaktykę zakrzepicy żył głębokich (ZŻG) i zatorowości płucnej (ZP). niższy przy nominalnym poziomie alfa 5% u pacjentów otrzymujących placebo w porównaniu z pacjentami otrzymującymi eteksylan dabigatranu.
Tabela 9: Przypadki krwawień w badaniu RE-SONATE, w którym oceniano profilaktykę zakrzepicy żył głębokich (ZŻG) i zatorowości płucnej (ZP)
* HR nie podlega ocenie, ponieważ w jednym z dwóch zabiegów nie ma zdarzenia
Definicja poważnych krwawień była zgodna z zaleceniami Międzynarodowego Towarzystwa Zakrzepicy i Hemostazy, jak opisano dla RECOVER i RECOVER II.
Atak serca
Zapobieganie udarom mózgu i zatorowości systemowej u dorosłych pacjentów z niezastawkowym migotaniem przedsionków (NAVF), z jednym lub większą liczbą czynników ryzyka
W badaniu RE-LY, w porównaniu z warfaryną, roczna częstość występowania zawału mięśnia sercowego dla eteksylanu dabigatranu wzrosła z 0,64% (warfaryna) do 0,82% (eteksylan dabigatranu 110 mg dwa razy na dobę) / 0,81% (eteksylan dabigatranu 150 mg dwa razy na dobę) ( patrz punkt 5.1).
Leczenie zakrzepicy żył głębokich (ZŻG) i zatorowości płucnej (ZP) oraz zapobieganie nawrotom ZŻG i ZP u dorosłych (ZŻG/ZP)
W trzech aktywnie kontrolowanych badaniach stwierdzono większą częstość występowania MI u pacjentów przyjmujących eteksylan dabigatranu niż u pacjentów przyjmujących warfarynę: 0,4% w porównaniu z 0,2% w krótkoterminowych badaniach RE-COVER i RE-COVER II; oraz 0,8% w porównaniu z 0,1% w długoterminowym badaniu RE-MEDY. W tym badaniu wzrost był istotny statystycznie (p = 0,022).
W badaniu RE-SONATE, w którym porównywano eteksylan dabigatranu z placebo, częstość występowania MI wynosiła 0,1% u pacjentów leczonych eteksylanem dabigatranu i 0,2% u pacjentów otrzymujących placebo (patrz punkt 4.4).
Populacja pediatryczna (ZŻG / ZP)
W badaniu klinicznym 1160,88 łącznie 9 nastoletnich pacjentów (wiek 12 z bólami brzucha; dyskomfort w jamie brzusznej) i 1 pacjent (11,1%) doświadczyło ciężkiego niepowiązanego zdarzenia niepożądanego (nawrót ŻChZZ w nodze) w okresie po leczeniu, więcej niż 3 dni po odstawieniu eteksylanu dabigatranu.
Zgłaszanie podejrzewanych działań niepożądanych
Zgłaszanie podejrzewanych działań niepożądanych występujących po dopuszczeniu produktu leczniczego do obrotu jest ważne, ponieważ umożliwia ciągłe monitorowanie stosunku korzyści do ryzyka produktu leczniczego. Osoby należące do fachowego personelu medycznego są proszone o zgłaszanie wszelkich podejrzewanych działań niepożądanych za pośrednictwem krajowego systemu zgłaszania. „adres https: //www.aifa.gov.it/content/segnalazioni-reazioni-avverse.
04.9 Przedawkowanie
Dawki eteksylanu dabigatranu wyższe niż zalecane narażają pacjenta na zwiększone ryzyko krwawienia.
W przypadku podejrzenia przedawkowania testy krzepnięcia mogą pomóc w określeniu ryzyka krwawienia (patrz punkty 4.4 i 5.1). Kalibrowany ilościowo test dTT lub powtarzane pomiary dTT pozwalają przewidzieć, kiedy zostaną osiągnięte określone poziomy dabigatranu (patrz punkt 5.1), nawet jeśli podjęte zostały inne środki, np. dializa.
Nadmierna aktywność przeciwzakrzepowa może wymagać przerwania leczenia produktem Pradaxa. Nie ma swoistego antidotum na dabigatran. W przypadku wystąpienia powikłań krwotocznych leczenie należy przerwać i zbadać przyczynę krwawienia Ponieważ dabigatran jest wydalany głównie przez nerki, należy utrzymać odpowiednią diurezę. Według uznania lekarza należy podjąć odpowiednie leczenie wspomagające, takie jak hemostaza chirurgiczna i przywrócenie objętości krwi.
Można rozważyć koncentraty aktywowanego kompleksu protrombiny (np. FEIBA) lub rekombinowanego czynnika VIIa lub koncentraty czynników krzepnięcia II, IX i X. Istnieją dowody eksperymentalne potwierdzające rolę tych leków w przeciwdziałaniu przeciwzakrzepowemu działaniu dabigatranu, ale dane dotyczące ich przydatności w warunkach klinicznych, a także możliwego ryzyka wystąpienia choroby zakrzepowo-zatorowej z odbicia są bardzo ograniczone. leków kontrastujących z efektem przeciwzakrzepowym. Należy zachować ostrożność podczas interpretacji wyników tych testów. Podanie koncentratów płytek krwi należy również rozważyć w przypadku wystąpienia małopłytkowości lub stosowania długo działających leków przeciwpłytkowych. Wszystkie leczenie objawowe należy stosować zgodnie z osądem lekarza.
W zależności od lokalnej dostępności, w przypadku dużego krwawienia, należy rozważyć konsultację z ekspertem ds. krzepliwości krwi.
Ponieważ wiązanie z białkami jest niskie, dabigatran można dializować; doświadczenie kliniczne wykazujące przydatność tego podejścia w badaniach klinicznych jest ograniczone (patrz punkt 5.2).
05.0 WŁAŚCIWOŚCI FARMAKOLOGICZNE
05.1 Właściwości farmakodynamiczne
Grupa farmakoterapeutyczna: leki przeciwzakrzepowe, bezpośrednie inhibitory trombiny.
Kod ATC: B01AE07.
Mechanizm akcji
Eteksylan dabigatranu jest drobnocząsteczkowym prolekiem, który nie wykazuje żadnej aktywności farmakologicznej. Po podaniu doustnym eteksylan dabigatranu jest szybko wchłaniany i przekształcany w dabigatran w wyniku hydrolizy katalizowanej esterazą w osoczu i wątrobie. Dabigatran jest silnym bezpośrednim, kompetycyjnym, odwracalnym inhibitorem trombiny i główną substancją czynną występującą w osoczu.
Ponieważ trombina (proteaza serynowa) umożliwia konwersję fibrynogenu do fibryny w kaskadzie krzepnięcia, jej hamowanie zapobiega tworzeniu się skrzepliny. Dabigatran hamuje wolną trombinę, trombinę związaną z fibryną i indukowaną przez trombinę agregację płytek.
Efekty farmakodynamiczne
Badania przeprowadzone na zwierzętach in-vivo I ex vivo wykazali skuteczność przeciwzakrzepową i działanie przeciwzakrzepowe dabigatranu po podaniu dożylnym oraz eteksylanu dabigatranu po podaniu doustnym w różnych modelach zwierzęcych zakrzepicy.
Istnieje wyraźna korelacja między stężeniem dabigatranu w osoczu a nasileniem działania przeciwzakrzepowego na podstawie danych z badań II fazy. Dabigatran wydłuża czas trombinowy (TT), ECT i aPTT.
Skalibrowany test czasu trombiny (dTT) dla dabigatranu w rozcieńczonym osoczu zapewnia oszacowanie stężenia dabigatranu w osoczu, które można porównać z oczekiwanymi stężeniami dabigatranu w osoczu.
ECT może stanowić bezpośrednią miarę aktywności bezpośrednich inhibitorów trombiny.
Test aPTT jest szeroko stosowany i daje przybliżone wskazanie intensywności efektu antykoagulacyjnego osiąganego przy użyciu dabigatranu, jednak test aPTT charakteryzuje się ograniczoną czułością i nie jest wskazany do dokładnej oceny ilościowej efektu antykoagulacyjnego, zwłaszcza przy wysokich stężeniach w osoczu przez dabigatran. Podwyższone wartości aPTT należy interpretować z ostrożnością.
Ogólnie rzecz biorąc, można argumentować, że te pomiary aktywności przeciwzakrzepowej odzwierciedlają poziomy dabigatranu i mogą stanowić wskazówki dotyczące oceny ryzyka krwawienia, tj. przekroczenie 90. percentyla poziomu dabigatranu w okresie minimalnym lub zmierzonego APTT w czasie leczenia uznaje się za powiązane ze zwiększonym ryzykiem krwawienia.
Prewencja pierwotna żylnych epizodów zakrzepowo-zatorowych w chirurgii ortopedycznej
W stanie stacjonarnym (po 3 dniach) średnia geometryczna stężenia dabigatranu w osoczu w czasie szczytowym, mierzona około 2 godziny po podaniu 220 mg eteksylanu dabigatranu, wynosiła 70,8 ng/ml, mieszcząc się w zakresie od 35 do 2-162 ng/ml ( 25-75 percentyl).
Średnia geometryczna stężenia dabigatranu w czasie minimalnym, mierzona pod koniec okresu dawkowania (tj. 24 godziny po podaniu dawki 220 mg dabigatranu), wynosiła średnio 22,0 ng/ml, w zakresie 13,0-35,7 ng/ml ( 25-75 percentyl).
U pacjentów leczonych w celu zapobiegania ŻChZZ po operacji wymiany stawu biodrowego lub kolanowego za pomocą 220 mg eteksylanu dabigatranu raz na dobę,
• 90. percentyl stężeń dabigatranu w osoczu, mierzony w czasie minimalnym (20-28 godzin po poprzedniej dawce), wynosił 67 ng/ml (patrz punkty 4.4 i 4.9),
• 90. percentyl aPTT w czasie minimalnym (20-28 godzin po poprzedniej dawce) wynosił 51 sekund lub 1,3-krotność górnej granicy normy.
U pacjentów leczonych w celu zapobiegania VTE po operacji wymiany stawu biodrowego lub kolanowego eteksylanu dabigatranu w dawce 220 mg raz na dobę nie wykonywano pomiarów EW.
Zapobieganie udarom i ES u dorosłych pacjentów z NLPZ z jednym lub więcej czynnikami ryzyka
W stanie stacjonarnym średnia geometryczna stężenia dabigatranu w osoczu w czasie szczytowym, mierzona około 2 godziny po podaniu 150 mg eteksylanu dabigatranu dwa razy na dobę, wynosiła 175 ng/ml, mieszcząc się w zakresie 117-275 ng/ml (25-75 percentyl). ). Średnia geometryczna stężenia dabigatranu w czasie minimalnym, mierzona rano pod koniec przerwy w dawkowaniu (tj. 12 godzin po wieczornej dawce 150 mg dabigatranu), wynosiła średnio 91,0 ng/ml, z przerwą równą 61,0- 143 ng / ml (25-75 percentyl).
U pacjentów z NVAF leczonych w celu zapobiegania udarom i SE za pomocą 150 mg eteksylanu dabigatranu dwa razy na dobę,
• 90. percentyl stężeń dabigatranu w osoczu, mierzony w czasie minimalnym (10-16 godzin po poprzedniej dawce), wynosił około 200 ng/ml,
• EW mierzony w czasie minimalnym (10-16 godzin po poprzedniej dawce), większy niż około 3-krotność górnej granicy normy odnosi się do 90. percentyla obserwowanego przy przedłużeniu EW o 103 sekundy,
• stosunek aPTT większy niż 2-krotność górnej granicy normy (wydłużenie aPTT o około 80 sekund) w czasie minimalnym (10-16 godzin po poprzedniej dawce) odzwierciedla 90. percentyl obserwacji.
Leczenie zakrzepicy żył głębokich (ZŻG) i zatorowości płucnej (ZP) oraz zapobieganie nawrotom ZŻG i ZP u dorosłych (ZŻG/ZP)
U pacjentów leczonych z powodu ZŻG i ZP eteksylanu dabigatranu w dawce 150 mg dwa razy na dobę średnia geometryczna stężenia dabigatranu w czasie minimalnym, mierzona w ciągu 10-16 godzin po podaniu dawki, pod koniec przerwy w dawkowaniu (tj. po 12 godzinach wieczorem 150 mg dabigatranu) wynosiła 59,7 ng/ml w zakresie 38,6-94,5 ng/ml (25-75 percentyl).W leczeniu ZŻG i ZP eteksylanu dabigatranu 150 mg dwa razy na dobę
• 90. percentyl stężeń dabigatranu w osoczu, mierzony w czasie minimalnym (10-16 godzin po poprzedniej dawce), wynosił około 146 ng/ml,
• EW mierzony w czasie minimalnym (10-16 godzin po poprzedniej dawce), większy niż około 2,3-krotność wartości wyjściowej odnosi się do 90. percentyla obserwowanego dla wydłużenia EW o 74 sekundy,
• 90. percentyl aPTT w czasie minimalnym (10-16 godzin po poprzedniej dawce) wynosił 62 sekundy lub 1,8-krotność wartości wyjściowej.
Brak danych farmakokinetycznych u pacjentów leczonych w celu zapobiegania nawrotom ZŻG i ZP za pomocą 150 mg eteksylanu dabigatranu dwa razy na dobę.
Skuteczność kliniczna i bezpieczeństwo
Pochodzenie etniczne
Nie zaobserwowano istotnych różnic międzyetnicznych między pacjentami rasy białej, Afroamerykańskiej, latynoskiej, japońskiej lub chińskiej.
Badania kliniczne dotyczące profilaktyki żylnej choroby zakrzepowo-zatorowej (ŻChZZ) po dużej operacji wymiany stawu
W 2 dużych, randomizowanych badaniach prowadzonych w grupach równoległych, z podwójnie ślepą próbą, potwierdzających dawkę, pacjenci zakwalifikowani do poważnej operacji ortopedycznej (jeden do zabiegu aloplastyki stawu kolanowego i jeden do chirurgii aloplastyki stawu biodrowego) byli leczeni produktem Pradaxa w dawce 75 mg lub 110 mg w ciągu 1- 4 godziny po zabiegu, a następnie 150 lub 220 mg raz dziennie, hemostazę oceniono jako prawidłową lub 40 mg enoksaparyny dzień przed zabiegiem, a więc codziennie.
W badaniu RE-MODEL (aloplastyka stawu kolanowego) czas leczenia wynosił 6-10 dni, a w badaniu RE-NOVATE (aloplastyka stawu biodrowego) 28-35 dni.W sumie poddano leczeniu odpowiednio 2076 (aloplastyka stawu biodrowego) kolano) i 3494 (przemiana stawu biodrowego) pacjentów.
Połączenie wszystkich epizodów ŻChZZ (w tym ZP, proksymalnej i dystalnej ZŻG, zarówno objawowej, jak i bezobjawowej wykrytej za pomocą rutynowej flebografii) oraz śmiertelność z jakiejkolwiek przyczyny były pierwszorzędowymi punktami końcowymi obu badań.
Połączenie wszystkich głównych epizodów ŻChZZ (w tym ZP, proksymalnej i dystalnej ZŻG, zarówno objawowej, jak i bezobjawowej wykrytej za pomocą rutynowej flebografii) oraz śmiertelności związanej z ŻChZZ było drugorzędowym punktem końcowym uznawanym za bardziej istotny klinicznie.
Wyniki obu badań wykazały, że działanie przeciwzakrzepowe produktu Pradaxa w dawce 220 mg i 150 mg było statystycznie nie mniejsze niż enoksaparyny na całkowitą ŻChZZ i śmiertelność z jakiejkolwiek przyczyny. Dawka 150 mg była nieco gorsza niż w przypadku enoksaparyny (tabela 10.) Lepsze wyniki zaobserwowano w przypadku dawki 220 mg, gdzie szacowana częstość występowania poważnych epizodów ŻChZZ była nieco lepsza niż w przypadku enoksaparyny (tabela 10).
Badania kliniczne przeprowadzono w populacji pacjentów w średnim wieku > 65 lat.
W badaniach klinicznych fazy 3 nie stwierdzono różnic w skuteczności i bezpieczeństwie między mężczyznami i kobietami.
Spośród populacji pacjentów uczestniczących w badaniach RE-MODEL i RE-NOVATE (5539 leczonych pacjentów) 51% miało współistniejące nadciśnienie tętnicze, 9% współistniejącą cukrzycę, 9% chorobę wieńcową, a 20% miało w wywiadzie niewydolność żylną . Żaden z tych stanów nie wpływał na działanie dabigatranu na zapobieganie ŻChZZ lub częstość krwawień.
Dane dotyczące głównego punktu końcowego dotyczącego ŻChZZ i śmiertelności związanej z ŻChZZ były jednorodne w odniesieniu do pierwszorzędowego punktu końcowego skuteczności i przedstawiono w Tabeli 10.
Dane dotyczące punktów końcowych dla całkowitej ŻChZZ i śmiertelności z jakiejkolwiek przyczyny przedstawiono w Tabeli 11.
Dane dotyczące punktów końcowych krwawienia, które uznano za poważne, wymieniono w Tabeli 12 poniżej.
Tabela 10: Analiza głównych śmiertelności związanych z ŻChZZ i ŻChZZ w okresie leczenia w badaniach chirurgii ortopedycznej RE-MODEL i RE-NOVATE
Tabela 11: Analiza całkowitej śmiertelności ŻChZZ i śmiertelności z jakiejkolwiek przyczyny w okresie leczenia w badaniach chirurgii ortopedycznej RE-NOVATE i RE-MODEL
Tabela 12: Epizody poważnych krwawień (ESM) po leczeniu w poszczególnych badaniach RE-MODEL i RE-NOVATE
Zapobieganie udarom i ES u dorosłych pacjentów z NLPZ z jednym lub więcej czynnikami ryzyka
Kliniczne dowody skuteczności eteksylanu dabigatranu pochodzą z badania Randomized Evaluation of Long-term anticoagulant therapy (RE-LY), wieloośrodkowego, międzynarodowego, randomizowanego badania w grupach równoległych, porównującego dwie dawki zaślepionego eteksylanu dabigatranu (110 mg i 150 mg). mg dwa razy na dobę) w porównaniu do otwartej próby warfaryny u pacjentów z migotaniem przedsionków i umiarkowanym lub wysokim ryzykiem udaru mózgu i SE. Głównym celem badania było ustalenie, czy eteksylan dabigatranu nie jest gorszy od warfaryny pod względem zmniejszenia częstości występowania złożonych punktów końcowych udaru i SE. Oceniono także przewagę statystyczną.
W badaniu RE-LY zrandomizowano łącznie 18 113 pacjentów, w średnim wieku 71,5 roku i średnim wyniku CHADS2 2,1.Populacja pacjentów składała się z 64% mężczyzn, 70% rasy kaukaskiej i 16% Azjatów.W przypadku pacjentów zrandomizowanych do leczenia warfaryną, średni procent czasu w zakresie terapeutycznym (TTR) (INR 2-3) wynosił 64,4% (mediana TTR 67%).
Badanie RE-LY wykazało, że eteksylan dabigatranu w dawce 110 mg dwa razy na dobę nie jest gorszy od warfaryny w zapobieganiu udarom i SE u osób z migotaniem przedsionków, przy zmniejszonym ryzyku ICH, krwawienia całkowitego i dużego krwawienia . Dawka 150 mg dwa razy na dobę istotnie zmniejsza ryzyko udaru niedokrwiennego i krwotocznego, zgonu naczyniowego, ICH i całkowitego krwawienia w porównaniu z warfaryną. Częstość występowania poważnych krwawień po tej dawce była porównywalna z warfaryną. Częstość występowania zawału mięśnia sercowego była nieznacznie zwiększona po podaniu eteksylanu dabigatranu 110 mg dwa razy na dobę i 150 mg dwa razy na dobę w porównaniu z warfaryną (ryzyko względne odpowiednio 1,29; p = 0,0929 i ryzyko względne 1,27; p = 0,1240) Przy lepszym monitorowaniu INR obserwowane korzyści ze stosowania eteksylanu dabigatranu w porównaniu ze spadkiem warfaryny.
Tabele 13-15 przedstawiają szczegóły kluczowych ustaleń w całej populacji:
Tabela 13: Analiza częstości występowania udaru mózgu lub SE (pierwszorzędowy punkt końcowy) w okresie badania RE-LY.
% odnosi się do rocznej częstości występowania zdarzenia
Tabela 14: Analiza częstości występowania udaru niedokrwiennego lub krwotocznego w okresie badania RE-LY.
% odnosi się do rocznej częstości występowania zdarzenia
Tabela 15: Analiza częstości występowania śmiertelności z jakiejkolwiek przyczyny lub śmiertelności z przyczyn sercowo-naczyniowych w okresie badania RE-LY.
% odnosi się do rocznej częstości występowania zdarzenia
Tabele 16-18 przedstawiają pierwotne wyniki skuteczności i punkt końcowy bezpieczeństwa w głównych podpopulacjach:
W przypadku pierwszorzędowego punktu końcowego, udaru mózgu i SE, nie zidentyfikowano podgrup (tj. wieku, masy ciała, płci, czynności nerek, pochodzenia etnicznego itp.) o innym współczynniku ryzyka niż warfaryna.
Tabela 16: Ryzyko względne i 95% CI udaru / SE według podgrup
W przypadku pierwszorzędowego punktu końcowego bezpieczeństwa – poważne krwawienie, wystąpiła interakcja między efektami leczenia a wiekiem.Ryzyko krwawienia wzrastało wraz z wiekiem w przypadku dabigatranu w porównaniu z warfaryną. Ryzyko względne było wyższe u pacjentów w wieku ≥ 75 lat. Jednoczesne stosowanie leków przeciwpłytkowych ASA lub klopidogrelu w przypadku eteksylanu dabigatranu i warfaryny w przybliżeniu podwaja częstość występowania ESM. Nie stwierdzono istotnej interakcji z efektami leczenia w podgrupach oceniających czynność nerek i punktację CHADS2.
Tabela 17: Ryzyko względne i 95% CI poważnych krwawień według podgrup
RELY-ABLE (wieloośrodkowe długoterminowe badanie kontynuacyjne leczenia dabigatranem u pacjentów z migotaniem przedsionków, którzy ukończyli badanie RE-LY)
Rozszerzenie badania RE-LY (RELY-ABLE) dostarczyło dodatkowych danych dotyczących bezpieczeństwa dla kohorty pacjentów, którzy nadal przyjmowali tę samą dawkę eteksylanu dabigatranu zgodnie z leczeniem przypisanym w badaniu RE-LY. Pacjenci kwalifikowali się do badania RELY-ABLE. jeśli nie przerwali na stałe badanego produktu leczniczego w czasie ostatniej wizyty w ramach badania RE-LY. Badanie LY, do przedłużonej obserwacji do 43 miesięcy po RE-LY (średni okres obserwacji RE-LY + RELY-ABLE, 4,5 roku) Do badania włączono 5897 pacjentów, co stanowi 49% pacjentów pierwotnie przydzielonych do grupy otrzymującej dabigatran eteksylat w badaniu RE-LY i 86% pacjentów kwalifikujących się do RELY-ABLE.
Podczas dodatkowych 2,5 roku leczenia w badaniu RELY-ABLE, przy maksymalnej ekspozycji dłuższej niż 6 lat (całkowita ekspozycja w badaniu RE-LY + RELY-ABLE), długoterminowy profil bezpieczeństwa eteksylanu dabigatranu został potwierdzony dla obu 110 mg bid i 150 mg bid. Nie zaobserwowano nowych danych dotyczących bezpieczeństwa.
Częstość występowania zdarzeń, w tym poważnych krwawień i innych krwawień, była zgodna z obserwowaną w badaniu RE-LY.
Populacja pediatryczna
Europejska Agencja Leków uchyliła obowiązek przedstawienia wyników badań produktu Pradaxa we wszystkich podgrupach populacji pediatrycznej w celu zapobiegania epizodom zakrzepowo-zatorowym w zatwierdzonym wskazaniu (informacje dotyczące stosowania u dzieci, patrz punkt 4.2).
Pochodzenie etniczne (profilaktyka udaru mózgu u pacjentów z migotaniem przedsionków)
Nie zaobserwowano istotnych różnic etnicznych między pacjentami rasy białej, Afroamerykańskiej, latynoskiej, japońskiej lub chińskiej.
Skuteczność kliniczna i bezpieczeństwo (leczenie ZŻG/ZP)
Leczenie zakrzepicy żył głębokich (ZŻG) i zatorowości płucnej (ZP) u dorosłych (leczenie ZŻG/ZP).
Skuteczność i bezpieczeństwo badano w dwóch wieloośrodkowych, randomizowanych badaniach z podwójnie ślepą próbą, prowadzonych w grupach równoległych, RE-COVER i RE-COVER II. W badaniach tych porównywano eteksylan dabigatranu (150 mg dwa razy na dobę) z warfaryną (cel INR 2 , 0-3,0 ) u pacjentów z ZŻG i (lub) ostrą ZP. Głównym celem tych badań było ustalenie, czy eteksylan dabigatranu nie jest gorszy od warfaryny pod względem zmniejszenia częstości występowania złożonego pierwszorzędowego punktu końcowego nawrotu ZŻG i (lub) objawowego ZP i związanego zgonów w ciągu 6 miesięcy leczenia.
W badaniach RE-COVER i RE-COVER II zrandomizowano 5153 pacjentów, a 5107 poddano leczeniu.
Czas trwania leczenia ustaloną dawką dabigatranu wynosił 174,0 dni bez kontroli krzepnięcia. W przypadku pacjentów zrandomizowanych do leczenia warfaryną mediana czasu w zakresie terapeutycznym (INR 2,0 do 3,0) wyniosła 60,6%.
Badania wykazały, że leczenie eteksylanem dabigatranu w dawce 150 mg dwa razy na dobę nie było gorsze od leczenia warfaryną (margines non-inferiority dla RE-COVER i RE-COVER II: 3,6 dla różnicy ryzyka i 2,75 dla współczynnika ryzyka).
Tabela 18: Analiza pierwszorzędowych i drugorzędowych punktów końcowych skuteczności (ŻChZZ obejmuje ZŻG i (lub) ZP) do końca okresu po leczeniu w badaniach RE-COVER i RE-COVER II
Pochodzenie etniczne (leczenie DVT / PE)
Nie zaobserwowano istotnych różnic etnicznych między pacjentami rasy białej, Afroamerykańskiej, latynoskiej, japońskiej lub chińskiej.
Populacja pediatryczna (leczenie ZŻG/ZP)
Europejska Agencja Leków odroczyła obowiązek przedstawienia wyników badań produktu Pradaxa we wszystkich podgrupach populacji pediatrycznej w leczeniu ZŻG/ZP (informacje dotyczące stosowania u dzieci, patrz punkt 4.2).
Farmakokinetykę i farmakodynamikę eteksylanu dabigatranu podawanego dwa razy na dobę przez trzy kolejne dni (w sumie 6 dawek) pod koniec standardowego leczenia przeciwzakrzepowego oceniano w otwartym badaniu dotyczącym bezpieczeństwa i tolerancji przeprowadzonym u 9 klinicznie stabilnych nastolatków (w wieku od 12 do
Skuteczność kliniczna i bezpieczeństwo (profilaktyka ZŻG/ZP)
Zapobieganie nawrotom zakrzepicy żył głębokich (ZŻG) i zatorowości płucnej (ZP) u dorosłych (zapobieganie ZŻG/ZP)
Przeprowadzono dwa randomizowane badania z podwójnie ślepą próbą w grupach równoległych u pacjentów wcześniej leczonych lekami przeciwzakrzepowymi. Do kontrolowanego warfaryną badania RE-MEDY włączono pacjentów już leczonych od 3 do 12 miesięcy i wymagających dodatkowego leczenia przeciwzakrzepowego, a do kontrolowanego placebo badania RE-SONATE włączono pacjentów leczonych przez 6 do 18 miesięcy inhibitorami witaminy K.
Celem badania RE-MEDY było porównanie bezpieczeństwa i skuteczności doustnego eteksylanu dabigatranu (150 mg dwa razy na dobę) z warfaryną (docelowy INR 2,0-3,0) w długotrwałym leczeniu i zapobieganiu nawrotom objawowej ZŻG i/lub ZP. W sumie zrandomizowano 2866 pacjentów, a 2856 poddano leczeniu. Czas trwania leczenia eteksylanem dabigatranu wynosił od 6 do 36 miesięcy (mediana 534,0 dni). W przypadku pacjentów zrandomizowanych do leczenia warfaryną mediana czasu w zakresie terapeutycznym (INR 2,0-3,0) wyniosła 64,9%.
Badanie RE-MEDY wykazało, że leczenie eteksylatem dabigatranu w dawce 150 mg dwa razy na dobę było równoważne warfarynie (margines non-inferiority: 2,85 dla współczynnika ryzyka i 2,8 dla różnicy ryzyka).
Tabela 19: Analiza pierwszorzędowych i drugorzędowych punktów końcowych skuteczności (ŻChZZ obejmuje ZŻG i (lub) ZP) do końca okresu po leczeniu w badaniu RE-MEDY
Celem badania RE-SONATE była ocena wyższości eteksylanu dabigatranu nad placebo w zapobieganiu nawrotom objawowej ZŻG i (lub) ZP u pacjentów, którzy zakończyli już od 6 do 8 miesięcy leczenia AVK. Terapia obejmowała 6 miesięcy. z eteksylanem dabigatranu 150 mg dwa razy na dobę bez konieczności monitorowania.
Badanie RE-SONATE wykazało, że eteksylan dabigatranu skuteczniej niż placebo zapobiegał nawrotom objawowych epizodów ZŻG/ZP, w tym zgonów z niewyjaśnionych przyczyn, przy zmniejszeniu ryzyka z 5,6% do 0,4% (względna redukcja ryzyka o 92% w oparciu o współczynnik ryzyka) podczas okres leczenia (p
Badanie obejmowało okres obserwacji obserwacyjnej trwający 12 miesięcy po zakończeniu leczenia. Po przerwaniu badania działanie leku utrzymywało się do końca okresu obserwacji, co wskazuje na utrzymanie się początkowego efektu leczenia dabigatranem. Nie zaobserwowano efektu odbicia. Pod koniec okresu obserwacji zdarzenia ŻChZZ u pacjentów leczonych eteksylanem dabigatranu wyniosły 6,9% w porównaniu z 10,7% w porównaniu z grupą placebo (współczynnik ryzyka 0,61 (95% CI 0,42,0; 88), p = 0,0082).
Tabela 20: Analiza pierwszorzędowych i drugorzędowych punktów końcowych skuteczności (ŻChZZ obejmuje ZŻG i (lub) ZP) do końca okresu po leczeniu w badaniu RE-SONATE
Pochodzenie etniczne (zapobieganie ZŻG/ZP)
Nie zaobserwowano klinicznie istotnych różnic etnicznych między pacjentami rasy kaukaskiej, Afroamerykańskiej, latynoskiej, japońskiej lub chińskiej.
Populacja pediatryczna (profilaktyka ZŻG/ZP)
Europejska Agencja Leków odroczyła obowiązek przedstawienia wyników badań produktu Pradaxa we wszystkich podgrupach populacji pediatrycznej w zapobieganiu ZŻG/ZP (informacje dotyczące stosowania u dzieci, patrz punkt 4.2).
Badania kliniczne dotyczące zapobiegania chorobom zakrzepowo-zatorowym u pacjentów z protezami zastawek serca
W badaniu II fazy oceniano eteksylan dabigatranu i warfarynę u łącznie 252 pacjentów, którzy przeszli częściowo niedawną operację mechanicznej zastawki (tj. zakwalifikowani podczas hospitalizacji) i częściowo mechaniczną operację zastawki serca przez ponad trzy miesiące. W przypadku eteksylanu dabigatranu obserwowano więcej zdarzeń zakrzepowo-zatorowych (głównie udar i objawową/bezobjawową zakrzepicę zastawek) oraz większą liczbę krwawień niż w przypadku warfaryny. U pacjentów bezpośrednio pooperacyjnych duże krwawienie objawiało się głównie krwotocznym wysiękiem osierdziowym, szczególnie u pacjentów, u których eteksylan dabigatranu rozpoczęto wkrótce (tj. w 3. dniu) po operacji protezy zastawki serca (patrz punkt 4.3).
05.2 Właściwości farmakokinetyczne
Po podaniu doustnym eteksylan dabigatranu jest szybko i całkowicie przekształcany w dabigatran, który jest aktywną postacią w osoczu. Główną reakcją metaboliczną jest rozszczepienie proleku eteksylanu dabigatranu poprzez hydrolizę katalizowaną przez esterazę do substancji czynnej dabigatranu. Całkowita biodostępność dabigatranu po doustnym podaniu produktu Pradaxa wynosi około 6,5%.
Po podaniu doustnym produktu Pradaxa zdrowym ochotnikom profil farmakokinetyczny dabigatranu w osoczu charakteryzuje się szybkim wzrostem stężeń w osoczu, przy czym Cmax osiągane jest 0,5-2,0 godziny po podaniu.
Wchłanianie
Badanie oceniające pooperacyjne wchłanianie eteksylanu dabigatranu, 1-3 godziny po zabiegu, wykazało stosunkowo powolne wchłanianie w porównaniu do tego obserwowanego u zdrowych ochotników, wykazując profil stężenia w osoczu w funkcji czasu bez wysokich maksymalnych stężeń w osoczu. Maksymalne stężenie w osoczu osiągane jest 6 godzin po podaniu w okresie pooperacyjnym z powodu takich czynników jak znieczulenie, niedowład jelit i efekty chirurgiczne, niezależnie od postaci doustnej produktu leczniczego. W kolejnym badaniu wykazano, że powolne i opóźnione wchłanianie następuje zwykle dopiero w dniu zabiegu chirurgicznego.W kolejnych dniach wchłanianie dabigatranu jest szybkie, a maksymalne stężenie w osoczu osiągane jest 2 godziny po podaniu leku.
Pokarm nie zmienia biodostępności eteksylanu dabigatranu, ale opóźnia czas osiągnięcia maksymalnego stężenia w osoczu o 2 godziny.
Gdy peletki są przyjmowane bez kapsułki HPMC (hydroksypropylometyloceluloza), biodostępność doustna może wzrosnąć o 75% w porównaniu z preparatem odniesienia z kapsułką. Dlatego integralność kapsułek HPMC musi być zawsze zachowana podczas stosowania klinicznego, aby uniknąć niezamierzonego zwiększenia biodostępności eteksylanu dabigatranu. Dlatego należy poinformować pacjentów, aby nie otwierali kapsułek i nie przyjmowali ich zawartości samodzielnie (np. posypywać jedzeniem lub wlewać do napoju) (patrz punkt 4.2).
Dystrybucja
Zaobserwowano niezależne od stężenia (34-35%) wiązanie dabigatranu z białkami osocza ludzkiego. Objętość dystrybucji dabigatranu 60-70 l przekracza objętość wszystkich płynów ustrojowych, co wskazuje na umiarkowaną dystrybucję dabigatranu w tkankach.
Cmax i pole pod krzywą stężenia w osoczu od czasu były proporcjonalne do dawki Stężenia dabigatranu w osoczu wykazywały dwuwykładniczy spadek ze średnim końcowym okresem półtrwania wynoszącym 11 godzin u zdrowych osób w podeszłym wieku. Po wielokrotnych dawkach zaobserwowano „końcowy okres półtrwania wynoszący około 12-14 godzin". Okres półtrwania był niezależny od dawki. Okres półtrwania jest wydłużony, jeśli czynność nerek jest zaburzona, jak pokazano w tabeli 21.
Biotransformacja
Metabolizm i wydalanie dabigatranu badano po podaniu pojedynczej dożylnej dawki radioaktywnego dabigatranu zdrowym mężczyznom. Po podaniu dożylnym radioaktywność pochodząca z dabigatranu była eliminowana głównie z moczem (85%). Oszacowano, że wydalanie z kałem wynosi 6% podanej dawki, a całkowity odzysk radioaktywności wynosił od 88 do 94% podanej dawki w ciągu 168 godzin od podania.
Dabigatran podlega koniugacji z wytworzeniem farmakologicznie czynnych acyloglukuronidów.Istnieją cztery izomery pozycyjne 1-O, 2-O, 3-O, 4-O acyloglukuronidów, z których każdy stanowi mniej niż 10% całkowitego dabigatranu w osoczu. Ślady innych metabolitów są wykrywalne jedynie bardzo czułymi metodami analitycznymi. Dabigatran jest wydalany głównie w postaci niezmienionej z moczem z szybkością około 100 ml/min, co odpowiada szybkości przesączania kłębuszkowego.
Populacje specjalne
Niewydolność nerek
W badaniach I fazy ekspozycja (AUC) na dabigatran po doustnym podaniu produktu Pradaxa jest około 2,7 razy większa u ochotników z umiarkowanymi zaburzeniami czynności nerek (CrCL między 30 a 50 ml/min) niż u osób bez zaburzeń czynności nerek.
U niewielkiej liczby ochotników z ciężką niewydolnością nerek (CrCL 10-30 ml/min) ekspozycja na dabigatran (AUC) była około 6 razy większa, a okres półtrwania około 2 razy dłuższy niż obserwowany w populacji bez niewydolności nerek. pkt 4.2, 4.3 i 4.4).
Tabela 21: Okres półtrwania całkowitego dabigatranu u zdrowych osób i osób z zaburzeniami czynności nerek.
Klirens dabigatranu metodą hemodializy zbadano u 7 pacjentów ze schyłkową przewlekłą niewydolnością nerek (ESRD) bez migotania przedsionków. Dializę prowadzono przy szybkości przepływu dializatu 700 ml/min, przez cztery godziny i przy szybkości przepływu krwi zarówno 200 ml/min, jak i 350-390 ml/min. Spowodowało to usunięcie odpowiednio 50% do 60% stężeń dabigatranu. Ilość substancji usuwanej przez dializę jest proporcjonalna do szybkości przepływu krwi do 300 ml/min. Aktywność przeciwzakrzepowa dabigatranu zmniejszała się wraz ze zmniejszaniem się stężeń w osoczu, a zależność farmakokinetyczna/farmakodynamiczna nie została zmieniona przez procedurę.
Mediana CrCL w badaniu RE-LY wynosiła 68,4 ml/min. Prawie połowa (45,8%) pacjentów RE-LY miała CRL > 50 -
Mediana CrCL w badaniu RE-COVER wynosiła 100,4 ml/min. 21,7% pacjentów miało łagodną niewydolność nerek (CrCL>50 - 80 ml/min. Podobne wartości CrCL stwierdzono w badaniu RE-COVER II.
Mediana CrCL w badaniach RE-MEDY i RE-SONATE wyniosła odpowiednio 99,0 ml/min i 99,7 ml/min. 22,9% i 22,5% pacjentów miało CRL > 50-
Starsi pacjenci
Specyficzne badania farmakokinetyczne fazy I przeprowadzone u osób w podeszłym wieku wykazały 40 do 60% wzrost AUC i ponad 25% Cmax w porównaniu z młodymi osobami.
Wpływ wieku na ekspozycję na dabigatran został potwierdzony w badaniu RE-LY przy wyższym stężeniu minimalnym wynoszącym około 31% u osób w wieku ≥ 75 lat i niższym stężeniu minimalnym wynoszącym około 22% u osób w wieku
Zaburzenia czynności wątroby
Nie stwierdzono zmiany ekspozycji na dabigatran u 12 pacjentów z umiarkowanymi zaburzeniami czynności wątroby (stopień B wg klasyfikacji Child Pugh) w porównaniu z 12 osobami z grupy kontrolnej (patrz punkty 4.2 i 4.4).
Masy ciała
Stężenia dabigatranu w okresie minimalnym były o około 20% mniejsze u pacjentów o masie ciała > 100 kg w porównaniu z pacjentami o masie ciała pomiędzy 50 a 100 kg. Większość pacjentów (80,8%) miała masę ciała ≥ 50 kg i
Rodzaj
Ekspozycja na substancję czynną w prewencji pierwotnej epizodów ŻChZZ była o około 40% do 50% większa u kobiet i nie zaleca się dostosowywania dawki.U pacjentek z migotaniem przedsionków ekspozycja na substancję czynną była o 30% wyższa niż w przypadku pacjenci płci męskiej zarówno w czasie podawania leku, jak i po podaniu. Nie zaleca się dostosowywania dawki (patrz punkt 4.2).
pochodzenie etniczne
Nie zaobserwowano istotnych różnic międzyetnicznych między pacjentami rasy białej, Afroamerykańskiej, Latynosami, Japończykami lub Chińczykami w zakresie farmakokinetyki i farmakodynamiki dabigatranu.
Interakcje farmakokinetyczne
Prolekowy eteksylan dabigatranu jest substratem transportera wypływowego P-gp, ale nie dabigatranu.Z tego powodu jednoczesne stosowanie z inhibitorami transportera P-gp (amiodaron, werapamil, klarytromycyna, chinidyna, donedaron, tikagrelor i ketokonazol) oraz z induktorami (ryfampicyną) (patrz punkty 4.2, 4.4 i 4.5).
Badania interakcji in vitro nie wykazały hamowania ani indukcji głównych izoenzymów cytochromu P450. Zostało to potwierdzone w badaniach in vivo przeprowadzonych na zdrowych ochotnikach, w których nie wykazano interakcji pomiędzy tym leczeniem a następującymi substancjami czynnymi: atorwastatyną (CYP3A4), digoksyną (interakcja z transporterem P-gp) i diklofenakiem (CYP2C9).
05.3 Przedkliniczne dane o bezpieczeństwie
Dane z badań nieklinicznych, wynikające z konwencjonalnych badań farmakologicznych dotyczących bezpieczeństwa, toksyczności po podaniu wielokrotnym i genotoksyczności, nie ujawniają żadnego szczególnego zagrożenia dla ludzi.
Działania obserwowane w badaniach toksyczności po podaniu wielokrotnym były spowodowane wzmocnionym działaniem farmakodynamicznym dabigatranu.
Wpływ na płodność kobiet w postaci zmniejszonej implantacji i zwiększonej utraty przedimplantacyjnej zaobserwowano przy dawkach 70 mg/kg (5-krotność poziomu ekspozycji osoczowej u pacjentów). Przy dawkach toksycznych dla matek (5 do 10 razy przekraczających poziom ekspozycji w osoczu pacjentek) u szczurów i królików obserwowano zmniejszenie masy ciała i żywotności płodu wraz ze wzrostem zmian u płodu. W badaniu przed- i pourodzeniowym zaobserwowano wzrost śmiertelności płodów po podaniu dawek toksycznych dla matek (dawka odpowiadająca czterokrotnie większej ekspozycji w osoczu niż obserwowana u pacjentów).
W badaniach toksyczności przez całe życie na szczurach i myszach nie było dowodów na potencjał rakotwórczy dabigatranu do maksymalnej dawki 200 mg/kg.
Dabigatran, aktywna cząsteczka mesylanu eteksylanu dabigatranu, utrzymuje się w środowisku.
06.0 INFORMACJE FARMACEUTYCZNE
06.1 Zaróbki
Zawartość kapsułki
• Kwas winowy
• guma arabska
• Hypromeloza
• Dimetikon 350
• Talk
• Hydroksypropyloceluloza
Kapsuła
• karagen
• Chlorek potasu
• Dwutlenek tytanu
• Indygo karmin (E132)
• Żółcień pomarańczowa (E110)
• Hypromeloza
Czarny tusz do drukowania
• Szelak
• Czarny tlenek żelaza (E172)
• Wodorotlenek potasu
06.2 Niezgodność
Nieistotne.
06.3 Okres ważności
Blister i butelka: 3 lata.
Po otwarciu butelki produkt leczniczy należy zużyć w ciągu 4 miesięcy.
06.4 Specjalne środki ostrożności przy przechowywaniu
Pęcherz
Przechowywać w oryginalnym opakowaniu w celu ochrony przed wilgocią.
Butelka
Przechowywać w oryginalnym opakowaniu w celu ochrony przed wilgocią.Przechowywać butelkę szczelnie zamkniętą.
06.5 Rodzaj opakowania bezpośredniego i zawartość opakowania
Opakowania zawierające 10 x 1, 30 x 1 lub 60 x 1 kapsułek twardych, opakowanie zbiorcze zawierające 3 opakowania po 60 x 1 kapsułek twardych (180 kapsułek twardych) oraz opakowanie zbiorcze zawierające 2 opakowania po 50 x 1 kapsułek twardych (100 kapsułek twardych)). . Dodatkowo opakowania zawierające 6 białych aluminiowych blistrów, podzielnych według dawki jednostkowej (60 x 1). Blister składa się z górnej warstwy aluminium pokrytego kopolimerami polichlorku winylu-polioctanu winylu (akrylany PVCAC) w kontakcie z produktem oraz dolnej warstwy aluminium pokrytego polichlorkiem winylu (PVC) w kontakcie z produktem.
Butelka polipropylenowa z zakrętką zawierająca 60 kapsułek twardych.
Nie wszystkie rozmiary opakowań mogą być wprowadzone na rynek.
06.6 Instrukcje użytkowania i obsługi
Stosując lek Pradaxa pakowany w blistry, należy przestrzegać następujących instrukcji:
• Kapsułkę twardą należy wyjąć z blistra, podnosząc folię aluminiową z tyłu.
• Kapsułki twardej nie wolno przepychać przez blister.
• Folię aluminiową blistra należy unosić tylko wtedy, gdy potrzebna jest kapsułka twarda.
Podczas używania kapsułek pakowanych w butelkę należy przestrzegać następujących instrukcji:
• Butelkę otwiera się przez naciśnięcie i przekręcenie zakrętki.
Niewykorzystany lek i odpady pochodzące z tego leku należy usunąć zgodnie z lokalnymi przepisami.
07.0 PODMIOT POZWOLENIA NA DOPUSZCZENIE DO OBROTU
Boehringer Ingelheim International GmbH
Bingera 173
D-55216 Ingelheim am Rhein
Niemcy
08.0 NUMER POZWOLENIA NA DOPUSZCZENIE DO OBROTU
UE / 1/08/442/005
038451050
UE / 1/08/442/006
038451062
UE / 1/08/442/007
038451074
UE / 1/08/442/008
038451086
UE / 1/08/442/014
038451148
UE / 1/08/442/015
UE / 1/08/442/018
09.0 DATA PIERWSZEGO ZEZWOLENIA LUB PRZEDŁUŻENIA ZEZWOLENIA
Data pierwszego zezwolenia: 18 marca 2008 r.
Data ostatniego przedłużenia: 17 stycznia 2013 r.
10.0 DATA ZMIAN TEKSTU
18 grudnia 2014