Choroba ta zawdzięcza swoją nazwę amerykańskiemu endokrynologowi, który ją odkrył: Fredericowi Crosby Bartterowi.Roczną zachorowalność szacuje się na 1/830 000.
Istnieje kilka wariantów zespołu Barttera, których transmisja, choć nadal autosomalna, może zmieniać się od recesywnej do dominującej w zależności od przypadku.
Jeśli zespół Barttera nie zostanie szybko zdiagnozowany i leczony, może poważnie zaburzyć rozwój, wzrost i jakość życia pacjenta. Ponadto w szczególnie ciężkich przypadkach oczekiwana długość życia ulega znacznemu skróceniu.
Proszę zanotować
Zespołu Barttera NIE należy mylić z zespołem Schwartza-Barttera, chorobą charakteryzującą się „upośledzonym wydzielaniem hormonu antyduiretowego (ADH), znanym również jako zespół nieprawidłowego wydzielania ADH (SIADH).
.zachodzące na poziomie pętli Henlego można przypisać „zmianie syntezy niektórych receptorów kanałowych/transporterowych (szczególnie białek transportujących jony o różnym charakterze) znajdujących się w tym” obszarze nerki. Zjawisko to jest spowodowane przez serię mutacji genetycznych wpływających na geny kodujące wyżej wymienione białka.
Różne warianty zespołu Barttera są rozróżniane w zależności od chorego genu. Więcej szczegółowych informacji na ten temat można znaleźć w następnym rozdziale.
.
Poniższa tabela przedstawia zatem różne warianty zespołu, zaangażowane zmutowane geny, białka (receptory/transportery kanałowe), które kodują, oraz obraz kliniczny danego wariantu.
Wariant
Zmutowany gen
Zaangażowany kanał/transporter
Prezentacja kliniczna
Zespół Barttera typu I
Gen SLC12A1
NKCC2 (kotransporter sodowo-potasowo-chlorowy lub Na+/K+/2Cl-)
Prenatalny (lub niemowlęcy) zespół Barttera
Zespół Barttera typu II
Gene KCNJ1
ROMK (kanał potasowy rdzenia zewnętrznego nerki)
Prenatalny (lub niemowlęcy) zespół Barttera
Zespół Barttera typu III
Gen CLNKb
CLCNKb (kanał chlorowy typu Kb)
Klasyczny zespół Barttera
Zespół Barttera typu IV lub IV A
Gene BSND
Barttina (podjednostka beta kanałów chlorowych typu Ka i Kb)
Prenatalny (lub niemowlęcy) zespół Barttera i niedosłuch odbiorczy
Zespół Barttera typ IV B
Geny CLCNKa i CLCNKb
CLCNKa (kanał chlorowy typu Ka) i CLCNKb
Prenatalny (lub niemowlęcy) zespół Barttera i głuchota czuciowo-nerwowa
Zespół Barttera typu V
gen CASR
CaSR (receptor wrażliwy na wapń)
Zespół Barttera z hipokalcemią
Jak widać z tabeli, pomimo obecności pięciu wariantów genetycznych, nie jest możliwe rozróżnienie tak wielu postaci klinicznych; w rzeczywistości wyróżnia się tylko cztery: prenatalny lub niemowlęcy zespół Barttera (typ I i II), klasyczny zespół Barttera (typ III), prenatalny lub niemowlęcy zespół Barttera związany z głuchotą czuciowo-nerwową (typ IV A i IV B; jednak niektóre źródła grupują te warianty razem z typem I i II) i wreszcie zespołem Barttera z hipokalcemią (typ V).
Czy wiedziałeś, że ...
Biorąc pod uwagę istnienie wariantu IV (lub IV A) i wariantu IV B zespołu Barttera, niektóre źródła rozważają łącznie sześć wariantów zespołu Barttera.Inne źródła uważają jednak wariant IV B za podtyp wariantu IV iz tego powodu rozważają istnienie tylko pięciu wariantów genetycznych zespołu Barttera.
Warianty typu I, II, III, IV i IV B są chorobami przenoszonymi autosomalnie recesywnie, co oznacza, że aby ujawnić się zespół, jednostka musi posiadać oba zmutowane allele dziedziczące je po rodzicach, którzy w związku z tym będą zdrowymi nosicielami. zespołu jest autosomalną dominującą chorobą transmisyjną, co oznacza, że do manifestacji objawów wystarczy, że pacjent posiada pojedynczy zmutowany allel, który w związku z tym może być dziedziczony nawet przez tylko jeden (również chory) ) obojga rodziców.
Pseudosyndrom Barttera
Pseudozespół Barttera jest stanem charakteryzującym się objawami podobnymi do tych wywoływanych przez zespół Barttera, ale którego przyczyną jest nadużywanie leków moczopędnych, takich jak furosemid.
Zespół Gitelmana
Zespół ten jest spowodowany zlokalizowaną mutacją genu SLC12A3, który koduje kotransporter chlorku sodu (NCC). W wyniku tej mutacji - przenoszonej w sposób autosomalny recesywny - u pacjenta dochodzi do upośledzenia resorpcji zwrotnej sodu, chloru i potasu na poziomie kanalika krętego dystalnego, w przeciwieństwie do zespołu Barttera, w którym upośledzenie resorpcji jest zlokalizowane w , zespół Gitelmana może dawać objawy podobne do zespołu Barttera, dlatego w praktyce klinicznej czasami trudno jest odróżnić te dwie choroby.
, hipochloremia i zasadowica metaboliczna, które mogą być związane z hiperreninemią (wysoki poziom reniny we krwi) i hiperaldosteronizmem. Oczywiście wszystkie te stany mogą z kolei powodować szereg objawów mogących pogorszyć jakość życia pacjenta (na przykład nudności, wymioty, zawroty głowy, osłabienie, ból głowy, niedociśnienie itp.).
Oprócz tego, co zostało powiedziane do tej pory, każdy wariant może wywołać określone objawy i objawy ściśle związane ze zmutowanym genem iw konsekwencji z zaangażowaniem kanału lub kotransportera, dla którego koduje ten gen. Dlatego poniżej zostaną pokrótce opisane typowe objawy i objawy związane z każdą z pięciu różnych postaci zespołu Barttera.
Zespół Barttera typu I
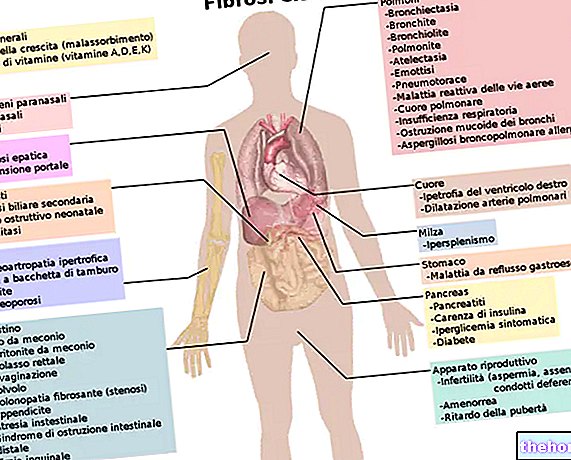
W zespole Barttera typu I mutacje wpływają na kodujący gen kotransportera sodowo-potasowo-chlorowego obecnego w pętli Henlego.W związku z upośledzonym wchłanianiem zwrotnym dochodzi do hipowolemii z powodu utraty soli. Jednocześnie, ponieważ reabsorpcja wapnia jest również powiązana z aktywnością wspomnianego kotransportera, jesteśmy świadkami wystąpienia hiperkalciurii, co może prowadzić do wystąpienia wapnicy nerek. Można również doświadczyć hipermagnezurii. W okresie prenatalnym może rozwinąć się wielowodzie wtórne do wielomoczu płodowego.
Zespół Barttera typu II
Zespół Barttera typu II jest spowodowany mutacją w genie kodującym kanał potasowy rdzenia nadnerczy. Manifestacje i objawy są podobne jak w wariancie I i również w tym przypadku można spotkać wielowodzie wtórne do wielomoczu płodowego. Jednak we wczesnym stadium noworodka może doświadczyć przemijającej hiperkaliemicznej kwasicy metabolicznej. Stan ten ewoluuje następnie w kierunku charakterystycznego obrazu klinicznego zespołu Barttera.
Zespół Barttera typu III
Znany również jako klasyczny zespół Barttera, wariant III choroby jest spowodowany mutacjami w genie kodującym kanał chlorowy typu Kb. Ponieważ kanały chlorowe typu Ka są zachowane w tej postaci, objawy wydają się być lżejsze, chociaż nadal obecne. Na ogół nie ma wapnicy nerek.
Zespół Barttera typu IV i IV B
W obu typach wariantu IV zaangażowane są geny zaangażowane w prawidłową syntezę kanałów chloru Ka i Kb.Ponieważ oba kanały są naruszone, objawy wydają się być bardziej nasilone niż w wariancie III zespołu. Niemowlęta mogą początkowo wykazywać obraz kliniczny naśladujący hipoaldosteronizm, który następnie ewoluuje w kierunku hipokaliemicznej zasadowicy metabolicznej, gdy organizm próbuje zrekompensować brak aktywności wspomnianych kanałów wapniowych. głuchota.
Zespół Barttera typu V
Wariant V zespołu Barttera jest spowodowany mutacją genu kodującego receptor wapniowy wrażliwy, biorący udział w hamowaniu reabsorpcji wody i różnych jonów, takich jak wapń, potas i sód.Receptor prowadzi do wystąpienia hipokalcemii i w konsekwencji hiperkalciuria związana z charakterystycznymi objawami zespołu Barttera.
Czy wiedziałeś, że ...
Warianty I, II, IV i IV B zespołu Barttera – jak również nazwa prenatalnego zespołu Barttera – są czasami określane również jako zespół hypeprostaglandyny E2, ponieważ charakteryzują się wzrostem stężenia tej prostaglandyny w osoczu.
- mające na celu określenie obecności i stężenia elektrolitów (sodu, potasu, chlorku, magnezu, wodorowęglanu, wapnia) oraz określonych substancji (reniny i aldosteronu) w osoczu i/lub moczu.Ostateczna diagnoza jest jednak możliwa tylko po wykonaniu określonych testów genetycznych.
Diagnozę różnicową należy natomiast postawić na podstawie pseudozespołu Barttera, zespołu Gitelmana, mukowiscydozy i celiakii.
W przypadkach, w których istnieje pewne ryzyko (na przykład rodzice ze zdrowymi i/lub chorymi nosicielami), że noworodek może objawić chorobę, możliwa jest również diagnostyka prenatalna.
z:
- Suplementy soli mineralnych (w szczególności, ale nie wyłącznie, potasu) w celu zrekompensowania braku reabsorpcji;
- Niesteroidowe leki przeciwzapalne (NLPZ), takie jak np. indometacyna Leki te podaje się w celu obniżenia nadmiernie wysokiego poziomu prostaglandyny E2;
- Leki moczopędne oszczędzające potas (podawane w celu zmniejszenia wydalania potasu z moczem).
W najpoważniejszych przypadkach i / lub w warunkach stresowych (wystąpienie innych chorób, interwencje chirurgiczne itp.) Uzupełnianie potasu i innych soli mineralnych można przeprowadzić dożylnie, oczywiście podobną operację musi przeprowadzić zdrowie wyspecjalizowany personel.